Creating Graphs
IGB can create several useful graphs from annotation tracks. Although the Depth graph can be utilized for all annotations, the usefulness shines, in combination with the Mismatch graph, with .bam and other short read alignment files. Here, you can see how many reads are present at each nucleotide position (Depth) and where mismatches occur as compared to the genomic sequence file.
In IGB 7.0 we had added the benefit that after these graphs are created, they will dynamically update if you load more data into the parent tracks. Just move around and hit Load Data; the tracks will update and so will the IGB track.
Graph tracks, such as these, can be saved as .wig formatted files.
Depth Graph
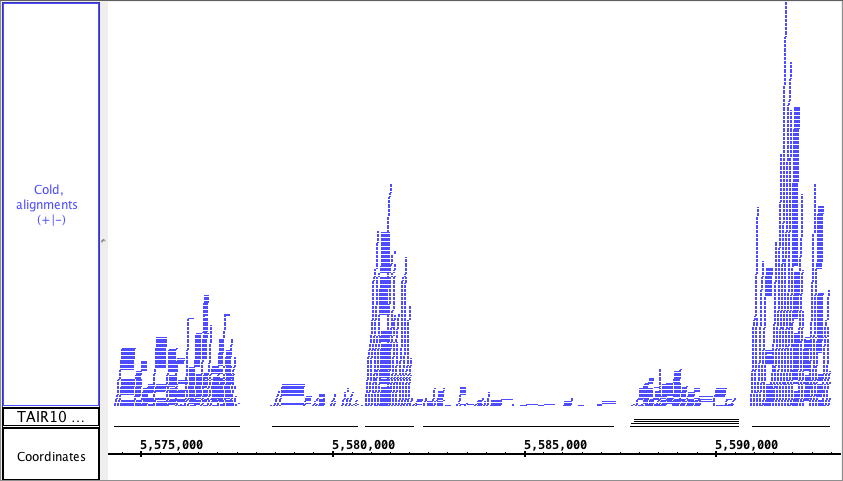
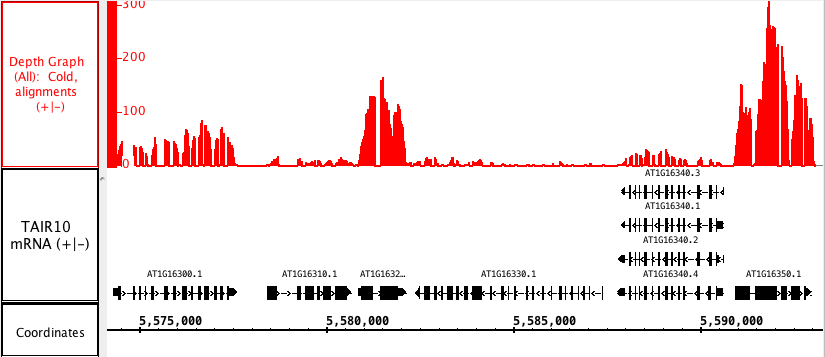
You can generate an overview of the relative density of annotation coverage across an entire chromosome. This feature dynamically graphs the number of annotations in a track that are present at each nucleotide across the genomic sequence. When you are completely zoomed in, and each pixel represents a single nucleotide base, the graph shows actual values for the number of reads aligned at that genomic location. Using the Select tool to hover over a particular point in the graph will open a tooltip with the actual number of reads at that point.
Right-click on the annotation in the label panel and choose Track Operations > Depth.
The picture below shows a fully expanded BAM file, showing the difficulty of seeing any details. The second picture shows just the depth graph (*BAM *track is 'hidden') with the tool tip showing the number of reads aligned onto that specific genomic location.
Mismatch Graph
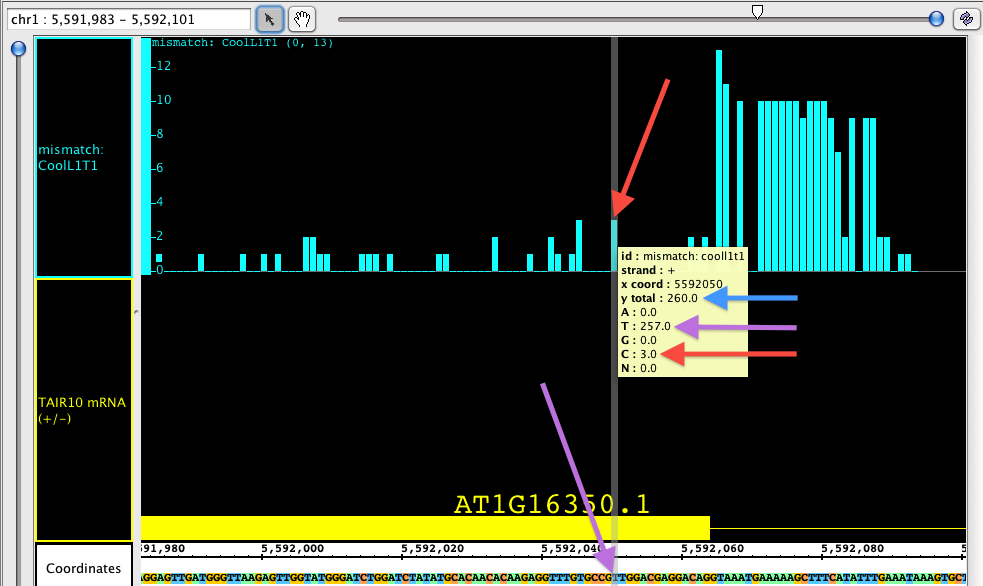
The Mismatch graph is specific to short read alignment files, such as BAM. The graph shows only the number of mismatched nucleotides across all reads at a specific genomic location, which can be very helpful in the detection of allelic variation, SNP identification and also error checking. Right click in the label to access the menu; choose Track Operations > Mismatch Graph. This function requires that the genomic sequence be loaded; if it is not loaded, IGB will load the sequence for you.
If you hover the Select tool (arrow cursor) over an individual bar of the graph, you will get a tool tip showing the total number of reads, and then the number of reads broken down by nucleotide. In the picture below, you can see that the total number of reads at the specified position is 260 (blue arrow). Of those reads, 257 at at 'T', which you can see is the matching nucleotide (purple arrows). 3 of those reads contain a 'G' instead; since G is a mismatch, the graph shows a bar with height of 3 (red arrows).
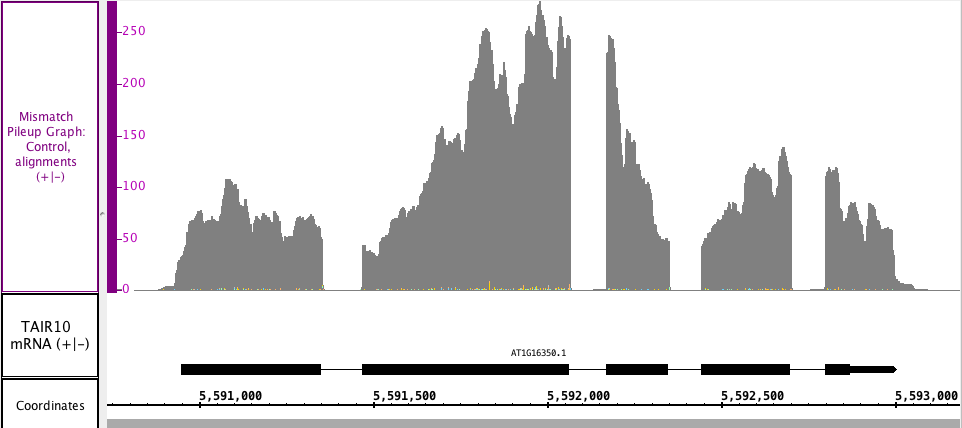
Mismatch Pileup Graph
In the Mismatch Pileup, you have the Depth portion represented as a dark gray graph, with the Mismatch overlayed in the matching nucleotide colors. The colors show which and how many of each nucleotide(s) are present at each mismatch position. The Mismatch Pileup graph, like the Mismatch graph, is also specific to short read alignment files, such as BAM. Right click in the label to access the menu; choose Track Operations > Mismatch Pileup Graph. This function requires that the genomic sequence be loaded; if it is not loaded, IGB will load the sequence for you.
In the image below, you can see the gray depth portion matches the Depth graph from above, but you can also see the mismatched nucleotides (very small numbers in this section), which are color coded by G, A, T and C.