...
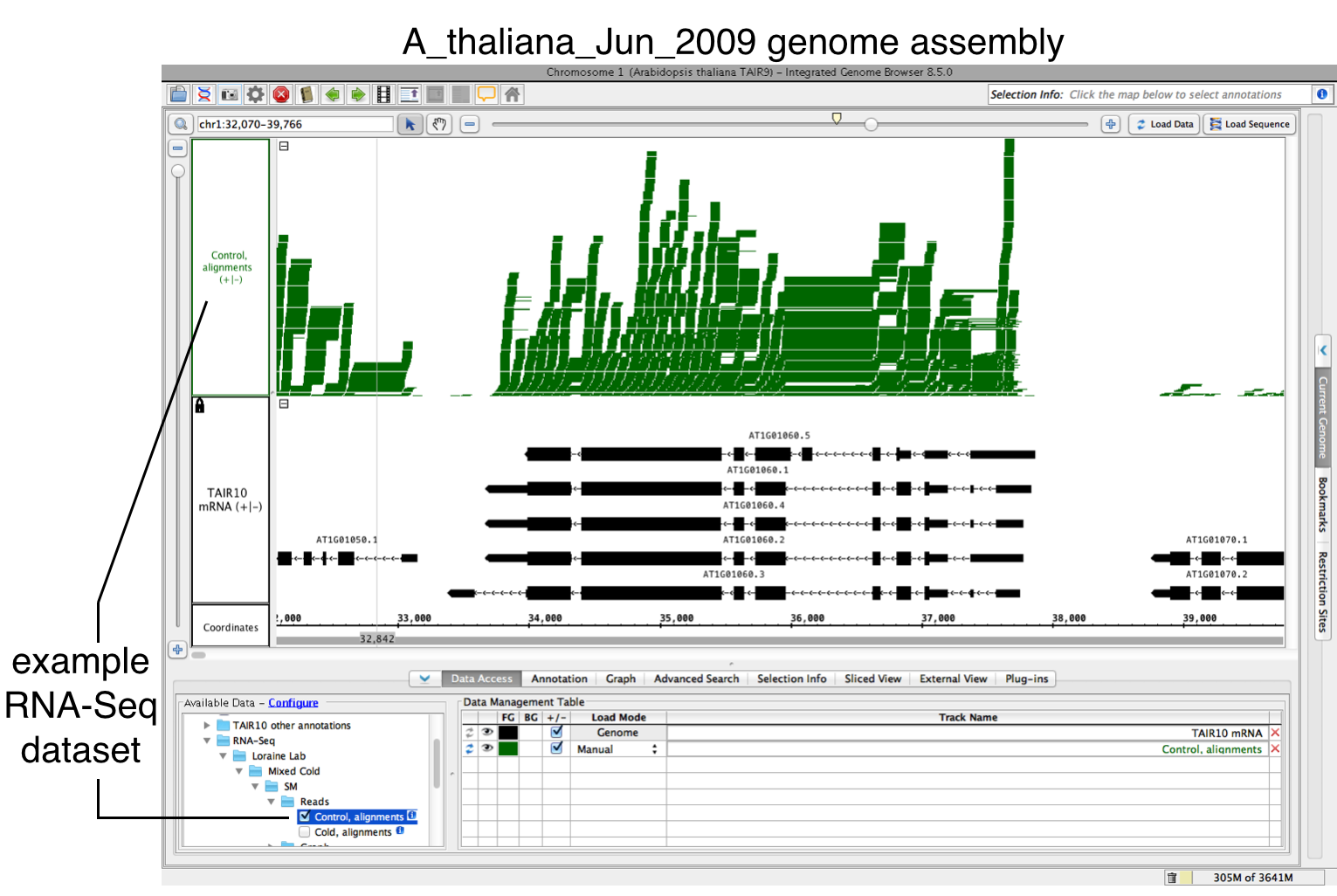
- Open a BAM file.
- Zoom in on the gene or region of interest.
- Click Load Data to load sequence read alignments into the new track.
To run FindJunctions:
- Right-click your data's track label.
- Select Track Operations > FindJunctions.Operations > FindJunctions or Track Operations > FindJunctions (TopHat).
- Enter a value or use the default. At least this many bases must align across a putative intron for a read to be counted as support for a junction.
- Select OK to run FindJunctions.
A new track will then appear containing junction features bracketing introns (see below). Labels report the number of spliced alignments that supported the junction. Each inferred intron appears as a thin line connecting two blocks, one on either side of the line. The width of these "flanking" blocks indicate the number of bases you entered in step 3 above, unless you selected the "Find Junctions (TopHat)" option. If you chose that option, then FindJunctions will create flanking blocks as large as the longest aligned region detected from any of the sequence read alignments. For example, if there was just one sequence that aligned across an inferred intron with 20 bases on either side of the intron, then the blocks will be 20 bases in size.
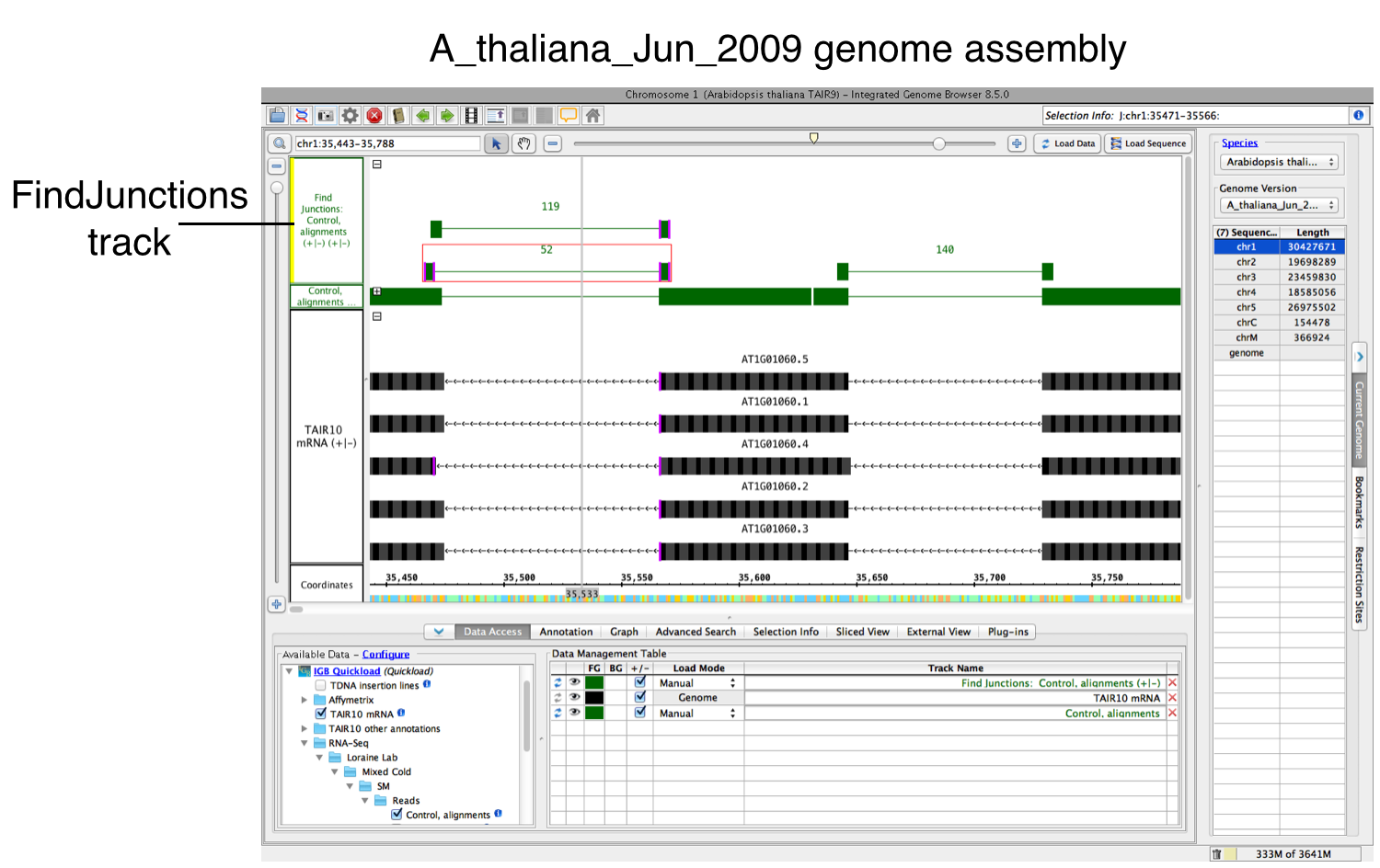
Find Junction
Using FindJunctions from the command line
...