Introduction
FindJunctions uses gapped RNA-Seq read to genome alignments to identify exon-exon junctions (i.e., introns). FindJunctions is implemented both as a visual analytics function within IGB and as a stand-alone, command-line program.
Within IGB, FindJunctions produces a new track showing exon-exon junction features labeled with the number of alignments that support that junction. The command-line program produces a BED file in which the score field contains the number of reads supporting a junction.
FindJunctions algorithm
FindJunctions operates on RNA-Seq read alignments, using alignments that contain gaps in the read sequence relative to the genomic sequence. These gaps in the read sequence correspond to introns and typically start and end with the so-called canonical splice site consensus sequences "GT" (5' end) and "AG" (3' end) for genes transcribed from the plus (forward) strand. For genes transcribed from the minus (reverse) strand, the consensus sequences relative to the plus strand are the reverse complement of the consensus splice site sequences (i.e., "CT" on the 5' end of the gap and "AC" on the 3' prime end).
For each alignment containing a gap, FindJunctions inspects the start and end coordinates of the gap and uses the genomic sequence to infer the strand, if available. FindJunctions creates a list of all such gaps, recording the strand and the genomic coordinates of the start and end coordinates. For each unique triplet of start, end, and strand, FindJunctions creates a scored junction feature and increments the score each time a gap supporting that feature is encountered in a dataset. Options are available to limit scoring to read alignments that have a given minimum number of bases flanking a gap and/or which having one unique mapping onto the genomic sequence.
How to use FindJunctions within IGB
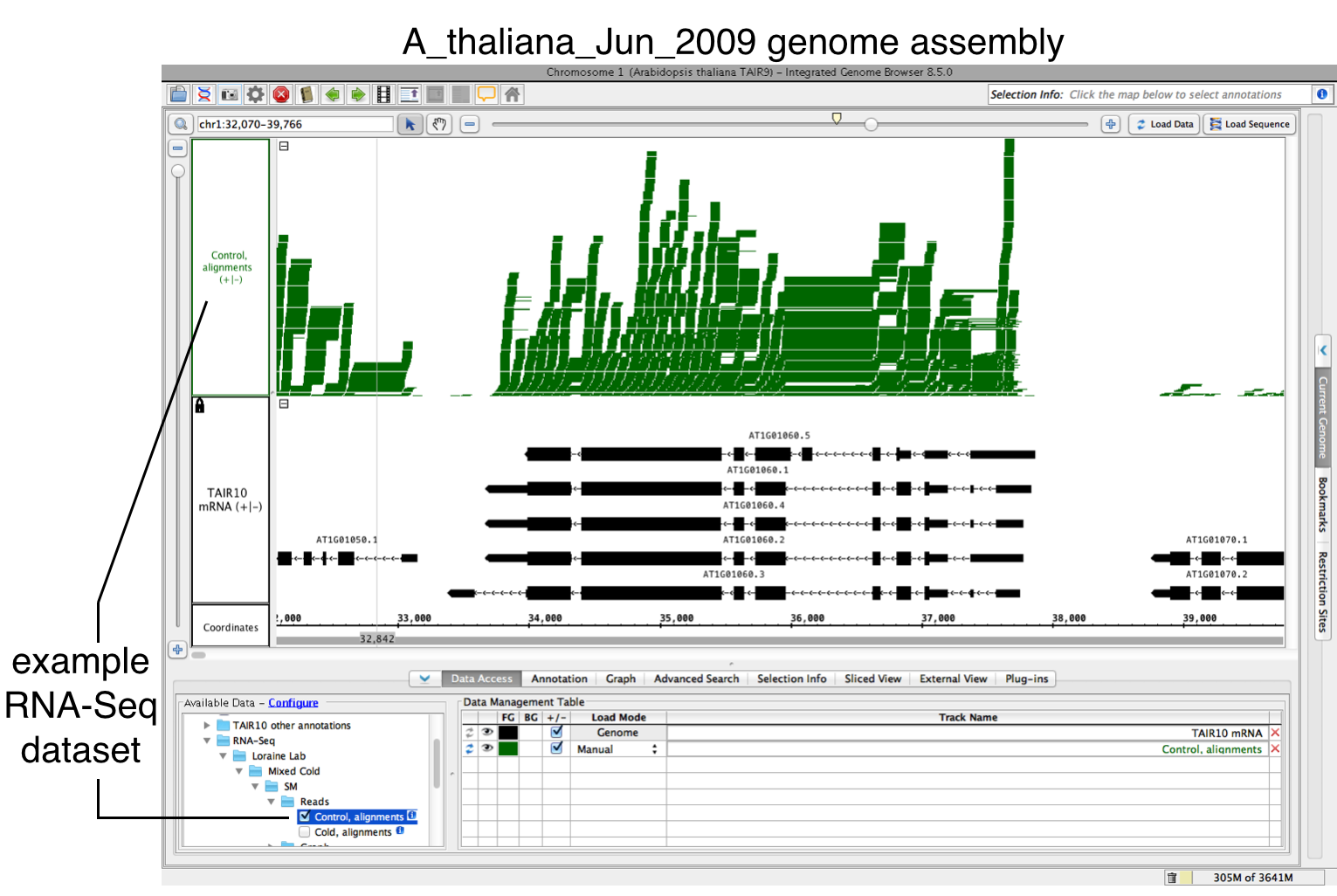
To use FindJunctions within IGB, you'll need to open and load your data, then run the FindJunctions track operation.
To open and load your data:
- Open a BAM file.
- Zoom in on the gene or region of interest.
- Click Load Data to load sequence read alignments into the new track.
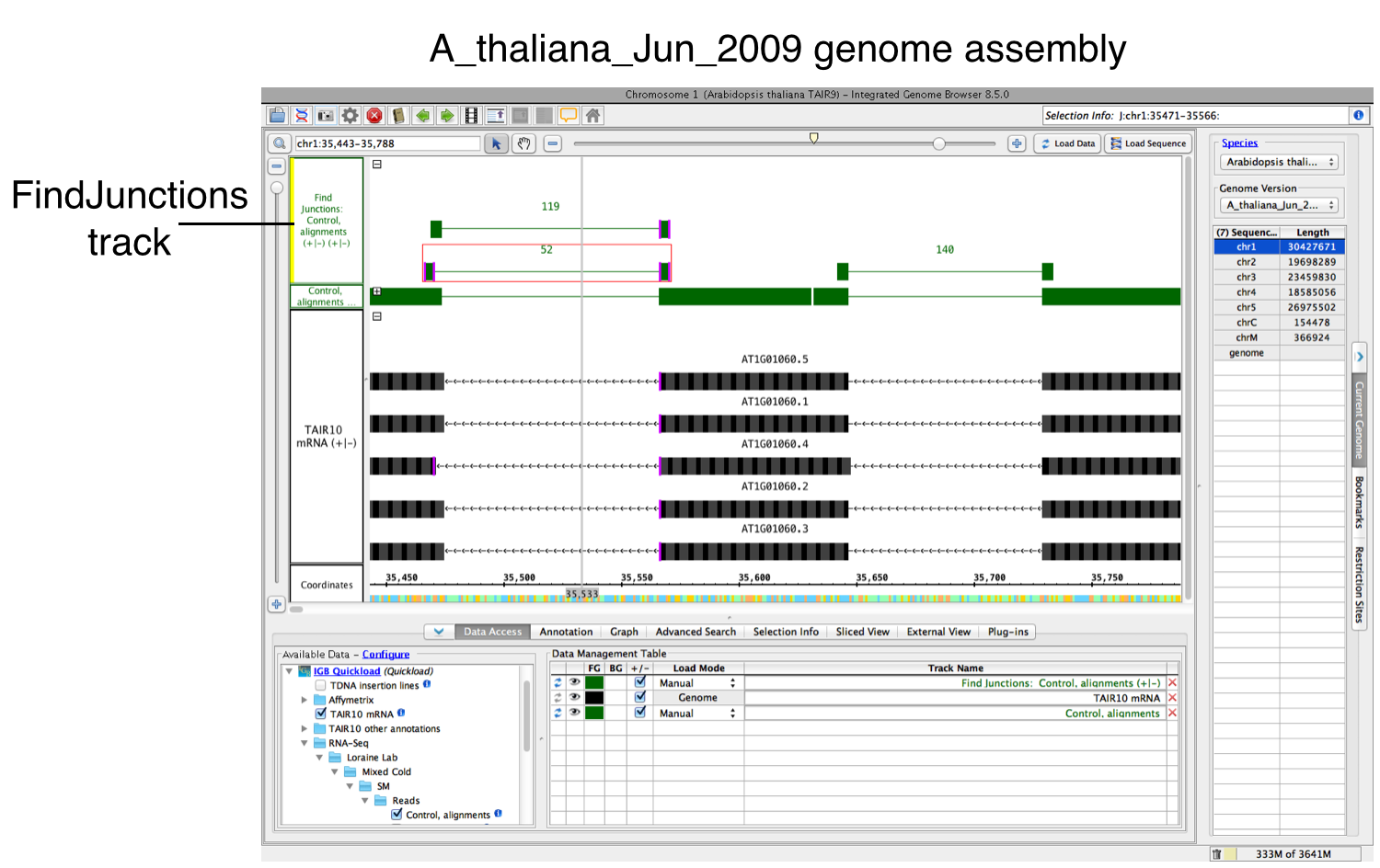
To run FindJunctions:
- Right-click your data's track label.
- Select Track Operations > FindJunctions or Track Operations > FindJunctions (TopHat).
- Enter a value or use the default. At least this many bases must align across a putative intron for a read to be counted as support for a junction.
- Select OK to run FindJunctions.

Did it work?
If yes, a new track will appear containing junction features bracketing introns (see below). Labels report the number of spliced alignments that supported the junction. Each inferred intron appears as a thin line connecting two blocks, one on either side of the line. The width of these "flanking" blocks indicate the number of bases you entered in step 3 above, unless you selected the "Find Junctions (TopHat)" option. If you chose that option, then FindJunctions will create flanking blocks as large as the longest aligned region detected from any of the sequence read alignments. For example, if there was just one sequence that aligned across an inferred intron with 20 bases on either side of the intron, then the blocks will be 20 bases in size. The name "TopHat" comes from a software program with similar behavior. TopHat is an RNA-Seq sequence to genomic assembly sequence alignment tool that created junction feature files (called "junctions.bed") using sequence read alignments. For more information about TopHat, visit the TopHat Manual.
Using FindJunctions from the command line
To run FindJunctions as a stand-alone program, visit https://bitbucket.org/lorainelab/findjunctions.
Follow the instructions there to compile FindJunctions and run it.