Follow these instructions if your genome is already supported in the main IGB QuickLoad site. To find out if your genome is already supported, see the instructions in Sharing data using QuickLoad sites.
Create a QuickLoad root directory.
Create a QuickLoad root directory (folder) on your local computer or on a Web server. This folder will contain your genome directories and a "meta-data" file called contents.txt.
Here is an example:
Create a directory for your genome version.
Create a genome directory corresponding to the genome assembly for which you want to share data via your QuickLoad. Like the main IGB QuickLoad site, you can support multiple genome versions from different species. Use the same names as in the main IGB QuickLoad site.
Here is an example:
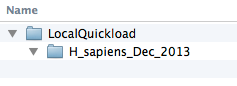
Create contents.txt file listing your genome.
Create a plain text file called contents.txt and save it your QuickLoad root directory. Copy the text for your genome from the main IGB QuickLoad contents.txt file into your file and save it.
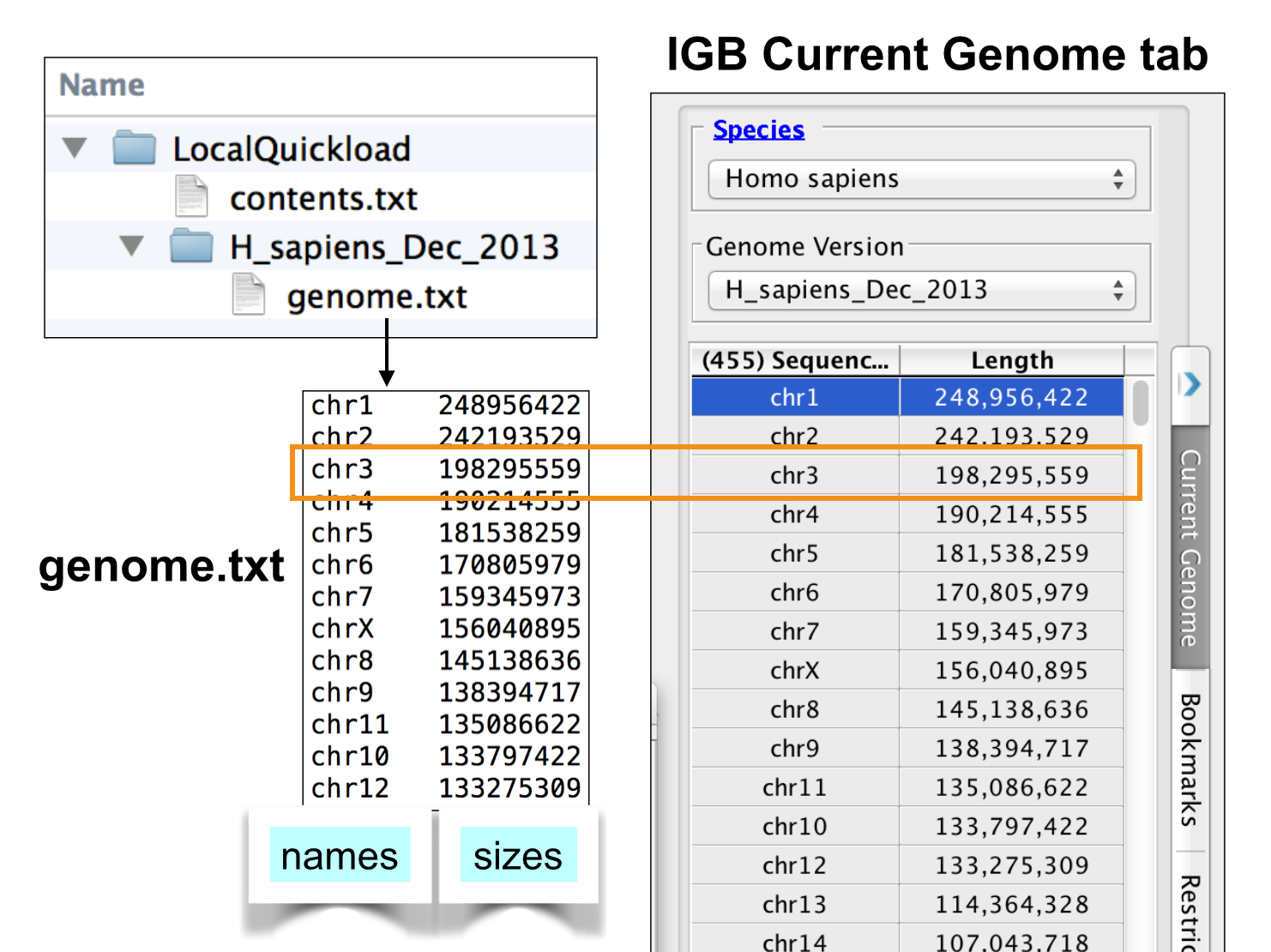
Add genome.txt file from public QuickLoad site.
Visit the public QuickLoad site in your Web browser and navigate to the directory for your genome version. Download the genome.txt from that directory and save it to your local genome version directory.
Note that IGB uses this file to create the sequence selection table under the Data Access Panel. The Data Access panel reports both sequence names and sizes, and so that's why the genome.txt file needs to contain both.
Create annots.xml meta-data file for each genome version directory.
Each genome version subdirectory needs an annots.xml file that is either empty or contains a listing of annotations you want to appear the Available Data Sets section of IGB's Data Access tabbed panel.
The annots.xml file is a simple "markup" file that specifies all the files that a user can load into IGB. Each file corresponds to a one track.
Here is a simple example:
<files> <file name="bamfiles/Treatment1.bam" title="RNA-Seq/Treatment Sample 1" description="RNA-Seq alignments from Treatment Sample 1" background="FFFFFF" foreground="0000FF" max_depth="15" name_size="12"/> </files>
The name attribute of the file tag indicates the physical location of the file relative to the directory where the annots.xml file resides. However, you can also use a URL to indicate the location of the file. This means that you the file is located on another server, you can leave it there; you don't have to copy it into your QuickLoad site.
For example, in this case, you could enter "http://www.example.com/bamfiles/Treatment1.bam" instead of "bamfiles/Treatmen1.bam" for file.
Note also that if you are hosting BAM files, the BAM file index ".bai" file needs to reside in the context root - i.e., the same directory - as the BAM file. If the index file is somewhere else, you can specify the location using the index attribute. This is useful if you are using cloud storage providers like Dropbox to serve data files.
For details on supported file attributes in an annots.xml file, see: About annots.xml
Add data files.
Place your annotation, BAM, CRAM, or graph (bedgraph or bigwig) files to the locations specified in your annots.xml file. You can you use any format IGB supports.
IGB QuickLoad can support BED, bedgraph, and GFF files that have been sorted, compressed, and indexed using the bgzip and tabix utilities. Doing this helps speed up data loading in IGB and allows loading data by region. For an example of how we used tabix to distribute coverage and junction files from an RNA-Seq data set, see Creating a new genome release for IGB QuickLoad from the IGB Developer's Guide.
Test your new QuickLoad site in IGB.
Tell IGB to use your new QuickLoad site.
Follow the directions in Adding and Managing Data Source Servers. To add the local server, click the "..." button and select the folder which contains the contents.txt file, your QuickLoad root directory.