| Table of Contents |
|---|
Introduction
Starting with IGB version 6.3, you can use IGB to visualize sequence alignment files alongside annotations and genome sequence.
...
| Note |
|---|
Each BAM file requires an index file (BAI) located in the same location (same folder, same website, etc.) as the BAM file. SAM files do not require indexes. |
Open short-read alignment (BAM or SAM) file
If your species and genome version of interest are available, first select it using menus in the Current Genome tab or click a species shortcut image on the IGB start screen.
...
| Info |
|---|
If your genome of interest is not available, you can enter it in the file chooser window. |
Set the load mode (optional)
Manual (the default)
When you first open a BAM file, its Load Mode will first be set to Manual, meaning that to load the data into IGB, you click the Load Data button. Also, only data for the currently shown region will load into IGB.
...
Since alignment data sets are typically very large, it is a good idea to only load a few genes' worth of data, depending on the file and the experiment.
Auto
The Auto load mode setting triggers automatic loading of data when the display is zoomed in past the Autoload threshold, indicated by the yellow marker on the horizontal zoom control.
...
To change the Autoload threshold to the current zoom level, choose View > Set Autoload Threshold to Current View
Zoom in on a region and load data
- Use the slider, double-click a gene model, run a search, or click-drag the sequence axis to zoom in.
- Click the Load Data button to load data into the display.
...
Read alignments (click to enlarge)
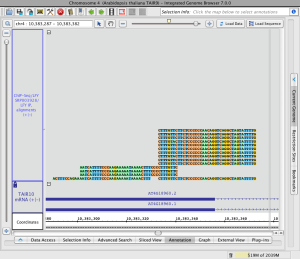
Load sequence data to view mismatches
To view just mismatches between reads and the reference genome
...
Read alignment with read sequence (click to enlarge)
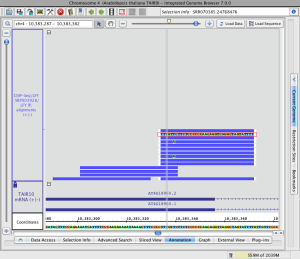
Adjust stack height
Note that in the image below showing read alignments from a ChIP-Seq experiment, there is a row of reads at the top of the track which appear darker than the rest. The darker row indicates there is some number of reads that are not being shown because they exceed the current stack height setting for the track.
...