General Function Checklist
Verify that all commonly used file formats are properly supported in IGB by loading all files available on the Smoke Testing Quickload.
- Add a Data Provider to IGB using the following URL: http://igbquickload.org/smokeTestingQuickload/
- The added Quickload site is available for the Homo sapien genome under the Available Data pane.
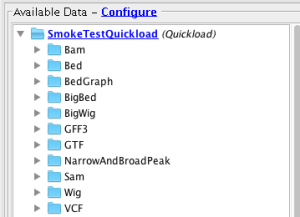
- Verify that all files in the Quickload site are loading into IGB (Data set names may differ from below - that's OK)
- Bam
- Bam_HomoSapien.bam
go to region: chr1:1,695,935-1,696,076
click Load Sequence to load sequence data and create track (gray), click Load Data to load data into it 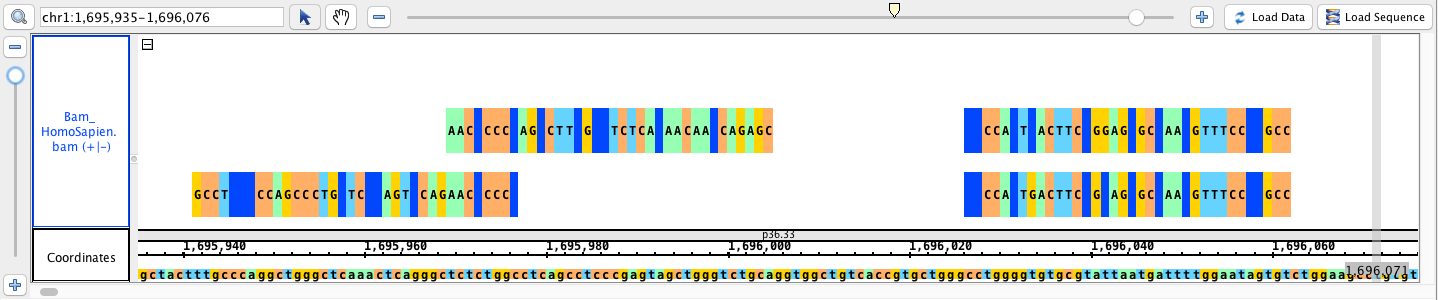
- Ensure soft-clipping can be hidden using the right-click menu, as shown below:
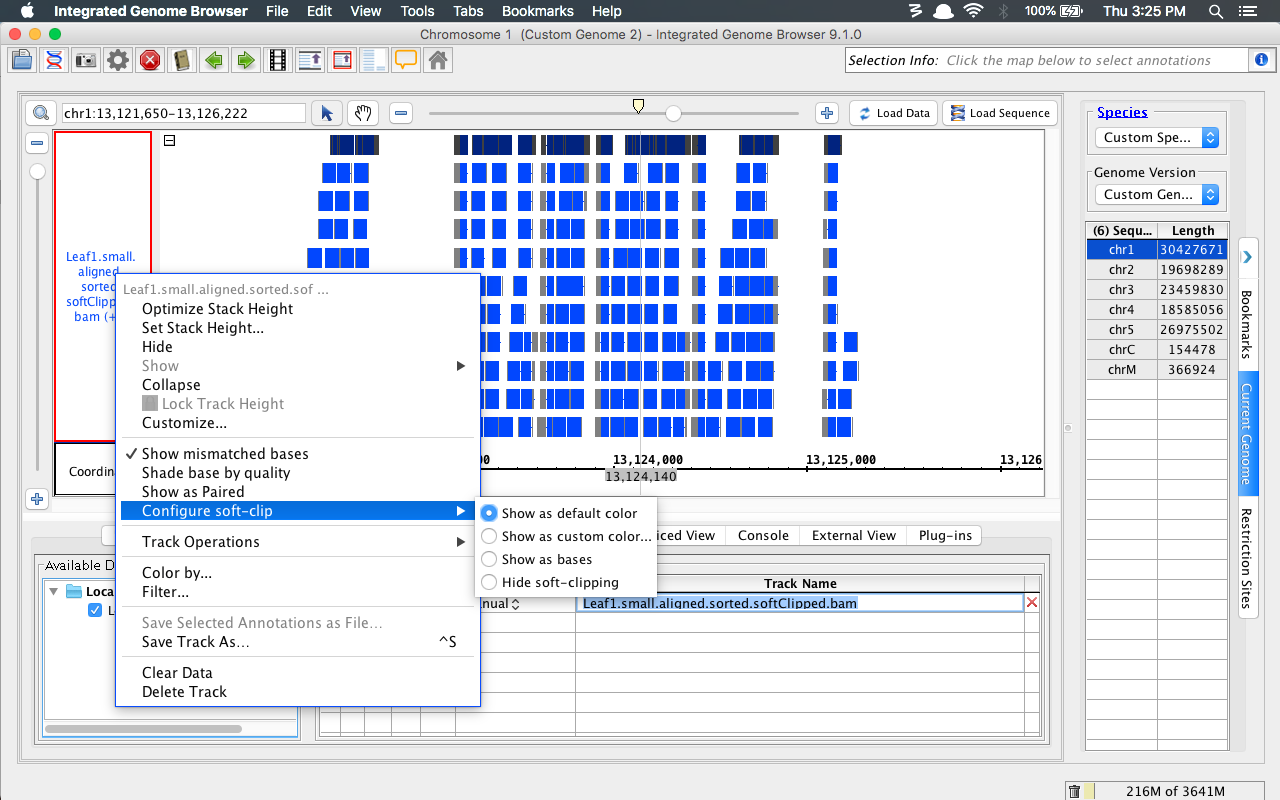
- Bam_HomoSapien.bam
- Bed
- Bed_HomoSapien.bed
- Bed_HomoSapien.bed.gz
region: chr1:1,699,059-1,912,174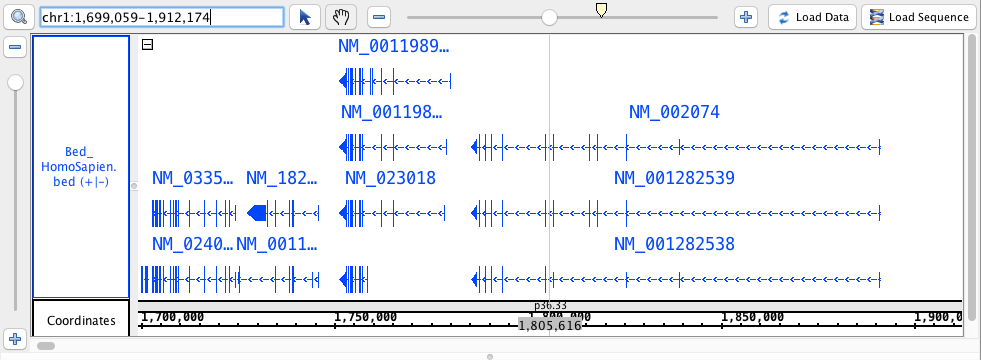
- BedGraph
- BedGraph_HomoSapien.bedgraph
- BedGraph_HomoSapien.bedgraph.gz
chr1:386,893-7,895,983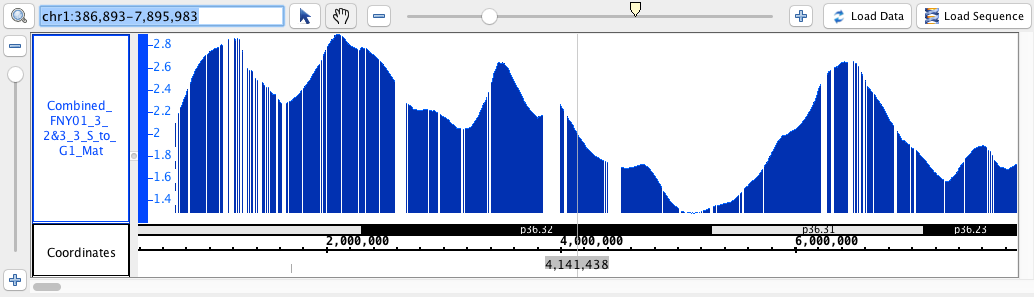
- BigBed
- BigBed_HomoSapien.bigbed
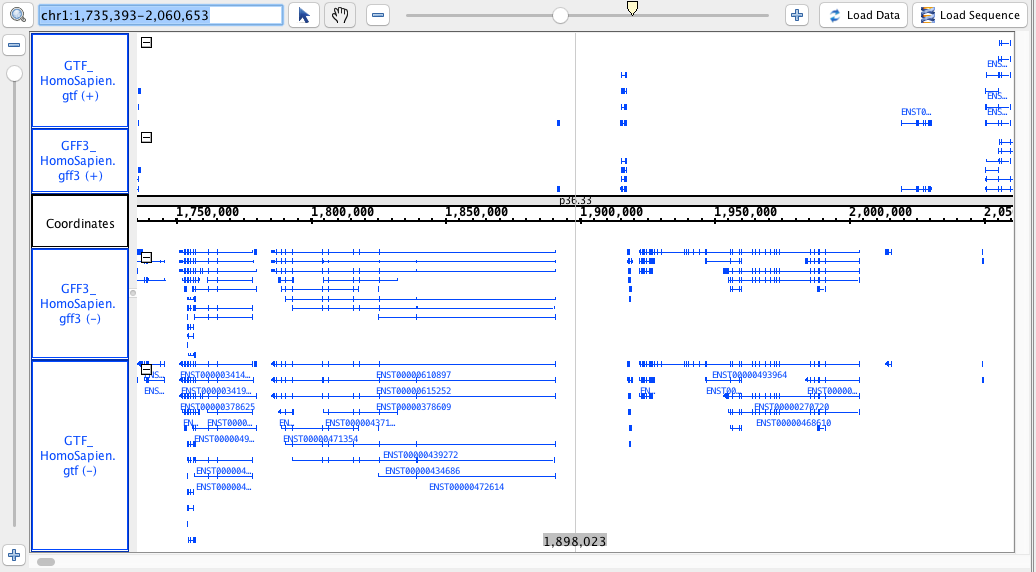
chr1:1,735,393-2,060,653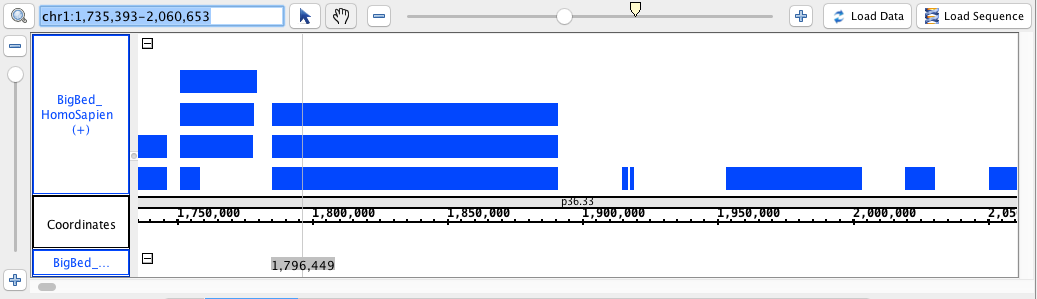
- BigBed_HomoSapien.bigbed
- BigWig
- BigWig_HomoSapien,bigwig
chr1:1,735,393-2,060,653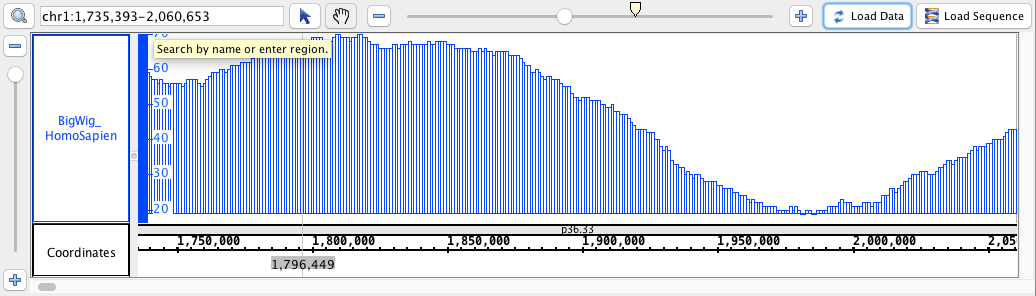
- BigWig_HomoSapien,bigwig
- GFF3
- GFF3_HomoSapien.gff3
- GFF3_HomoSapien.gff3.gz
region: chr1:1,735,393-2,060,653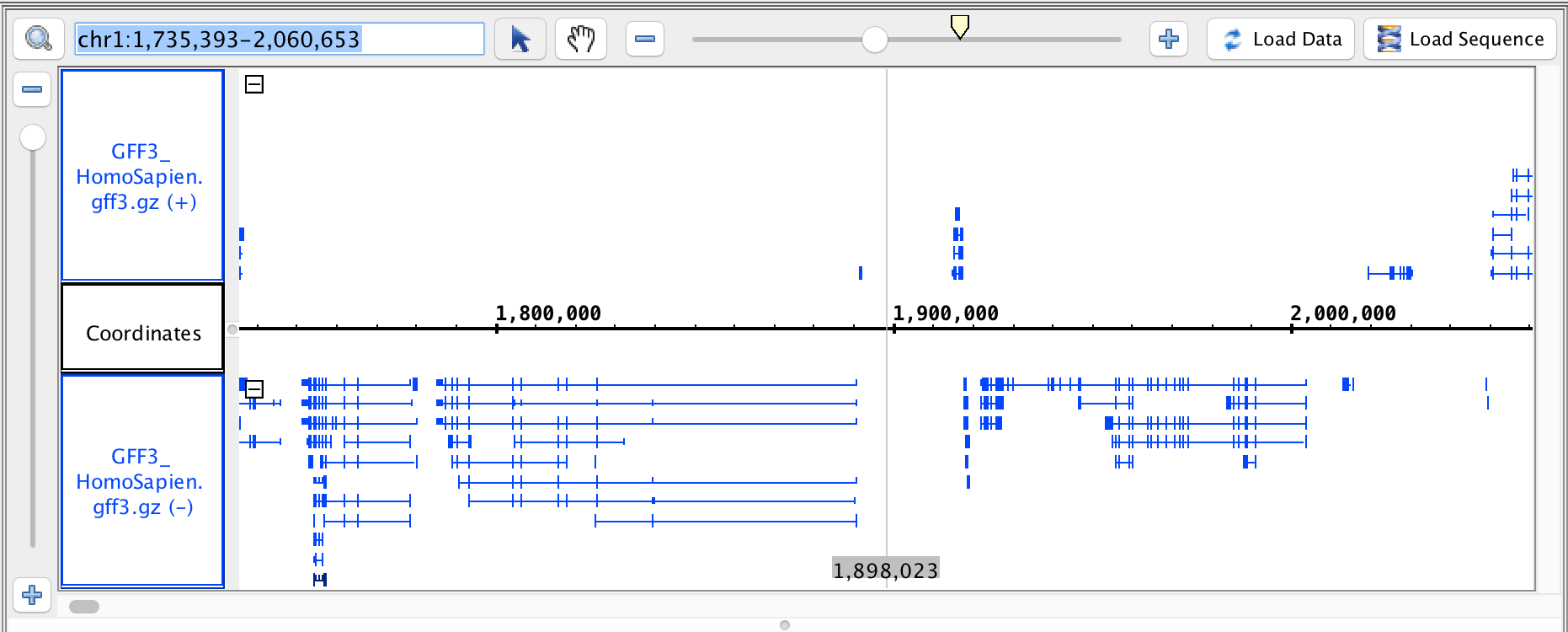
- Bam
- The added Quickload site is available for the Homo sapien genome under the Available Data pane.
- Add a Data Provider to IGB using the following URL: http://igbquickload.org/smokeTestingQuickload/
- GTF
- GTF_HomoSapien.gtf
- GTF_HomoSapien.gtf.gz
- NarrowAndBroadPeak
- BroadPeak_HomoSapien.broadpeak
- BroadPeak_HomoSapien.broadpeak.gz
chr1:800,118-800,619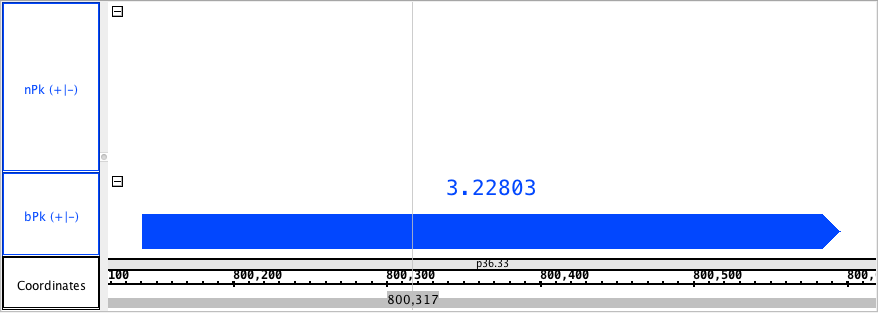
- NarrowPeak_HomoSapien.narrowpeak
- NarrowPeak_HomoSapien.narrowpeak.gz
chr1:9,361,070-9,361,207
chr1:0-12,643,524
- Sam
- Sam_HomoSapien_withHeader.sam
go to region: chr1:1,695,935-1,696,076
To get the dark blue blocks you will need to load the sequence.
The data in this file is identical to the Bam file above (Bam_HomoSapien.bam), it should appear identical to that.
- Sam_HomoSapien_withHeader.sam
- VCF
- VCF_HomoSapien.vcf
chr1:1,234,555-1,234,601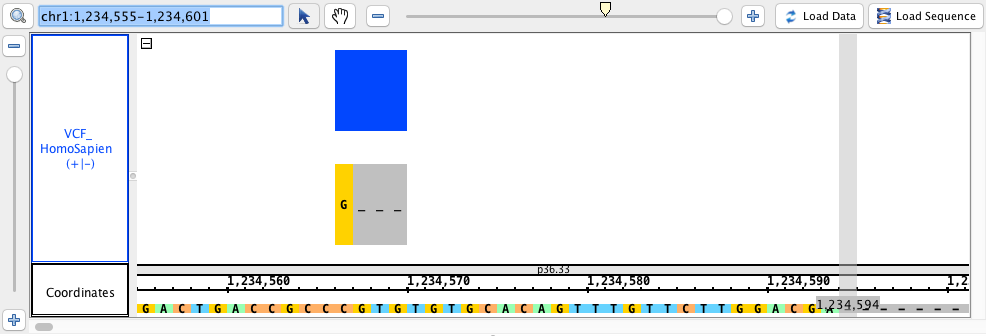
- VCF_HomoSapien.vcf
- Wig
- Wig_HomoSapien.wig
- Wig_HomoSapien.wig.gz
chr1:37,750,880-38,042,899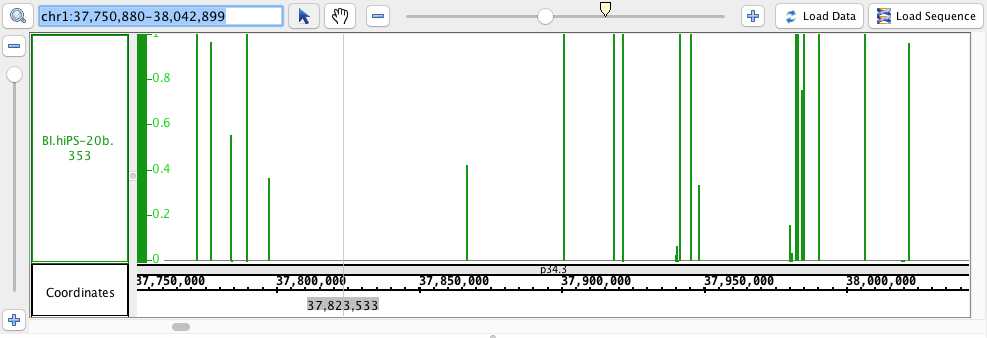
- GTF
Sequence appearance
Go to the Arabidopsis genome. From the RNA-Seq quicklaod (available by default as of 9.0.1), open the file RNA-Seq / Pollen SRP022162 / Reads / Pollen alignments. (url http://lorainelab-quickload.scidas.org/rnaseq/A_thaliana_Jun_2009/SRP022162/Pollen.bam)
Go to location: Chr1:7,313,640-7,319,896
Load Data and Load Sequence. Make sure the +/- option is checked for both the TAIR10_mRNA track (loaded by default) and the Pollen alignments track.
Go to location: Chr1:7,319,301-7,319,375
Optimize the stack height, and compare the main view to this image:
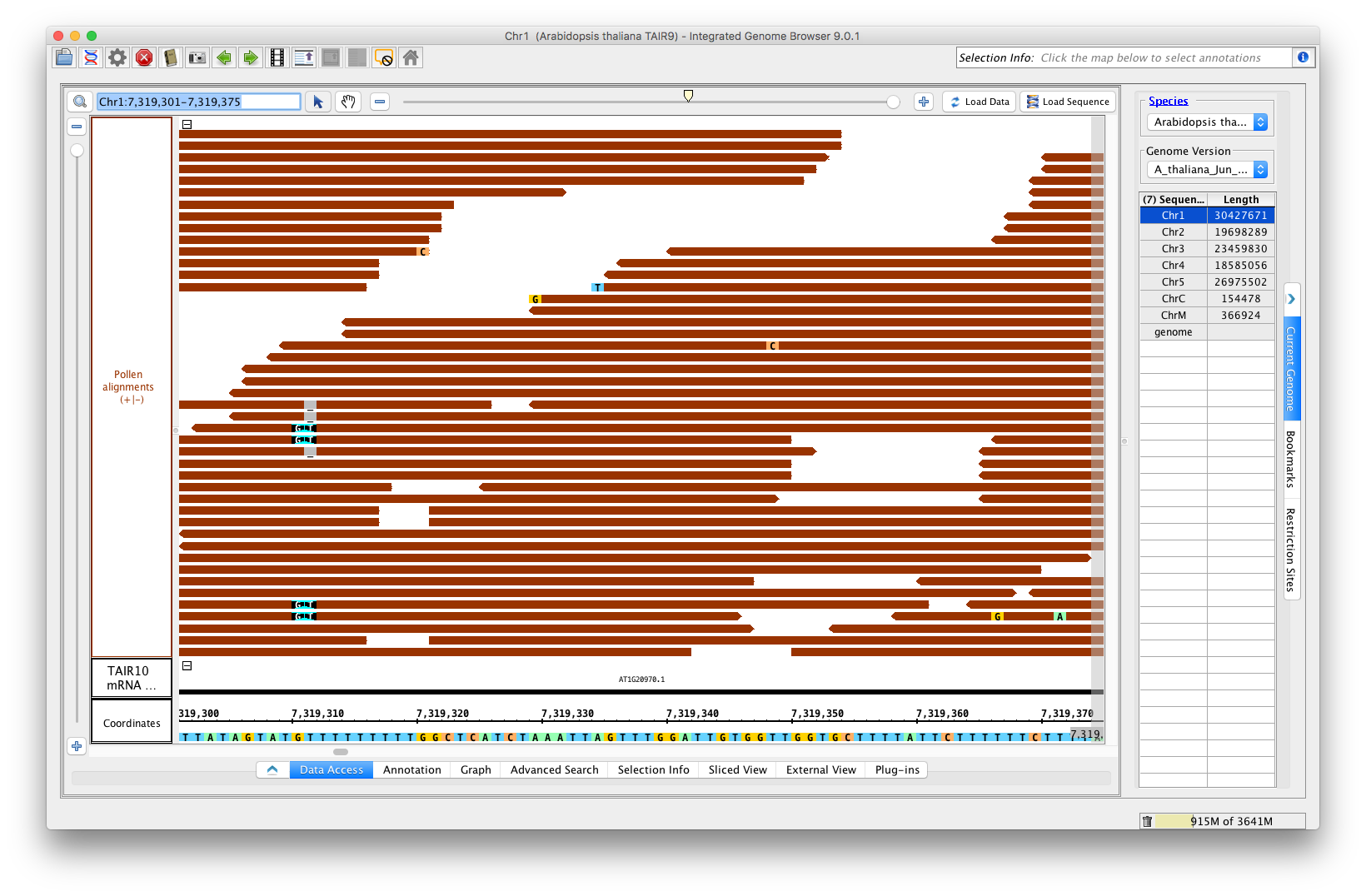
Verify that:
- single base mismatches of A, T, C, and G appear with a color scheme that matches the sequence in the coordinate axis (the bottom).
- deletions appear as shown, a gray space with a dash.
- insertions appear as shown (see position 7,319,310 in image)