General Function Checklist
Verify that all commonly used file formats are properly supported in IGB by loading all files available on the Smoke Testing Quickload.
- Add a Data Provider to IGB using the following URL: https://quickload-testing.s3.amazonaws.com/smokeTestingQuickload/
- The added Quickload site is available for the Homo sapien genome under the Available Data pane.
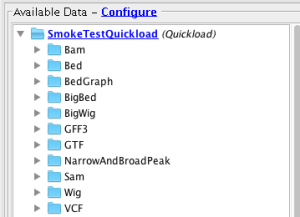
- Verify that all files in the Quickload site are loading into IGB (Data set names may differ from below - that's OK)
- Bam
- Bam_HomoSapien.bam
go to region: chr1:1,695,935-1,696,076
click Load Sequence to load sequence data and create track (gray), click Load Data to load data into it 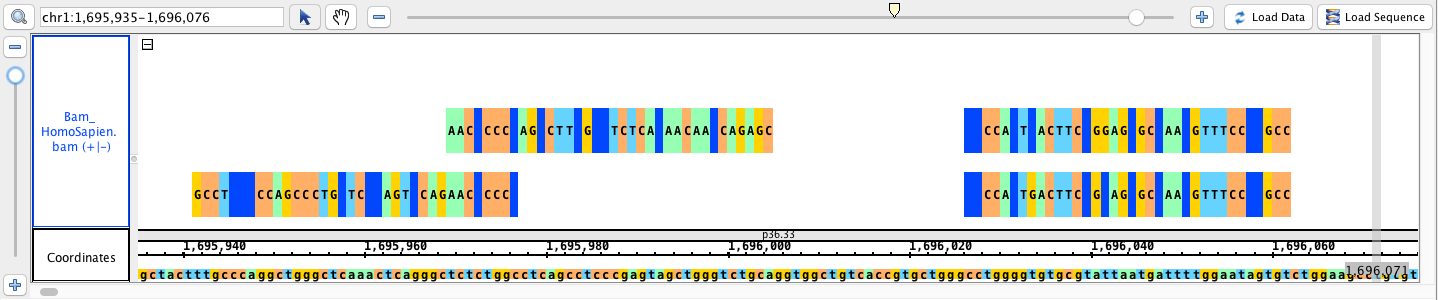
- Bam_HomoSapien.bam
- Bam - PacBio with softclipping
- pacBio.bam
chr1:9,971-10,034
click Load Sequence to load sequence data and create track (gray), click Load Data to load data into it
* note: sequence reads are imported in variable order, so your view may differ from the image below. Simply ensure that the gray portion of the reads
is consistently on the left side and generally aligns to the gray portion of the reference sequence (the NNNN portion) at the bottom, as well as that no
artifacts are present in your view that are not in the image below. 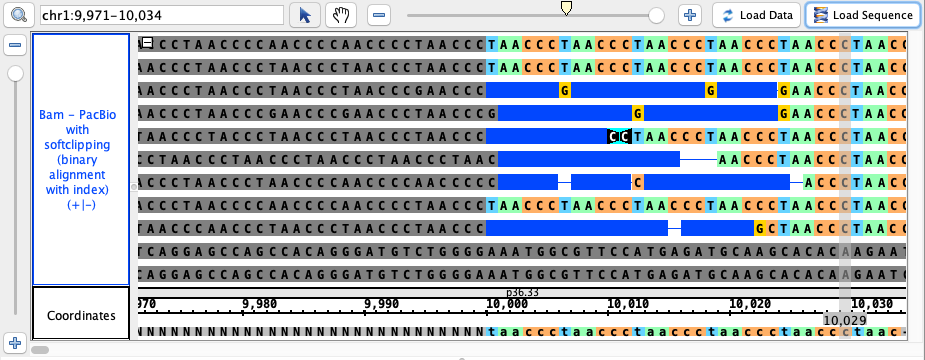
- pacBio.bam
- Bed
- Bed_HomoSapien.bed
- Bed_HomoSapien.bed.gz
chr1:1,699,059-1,912,174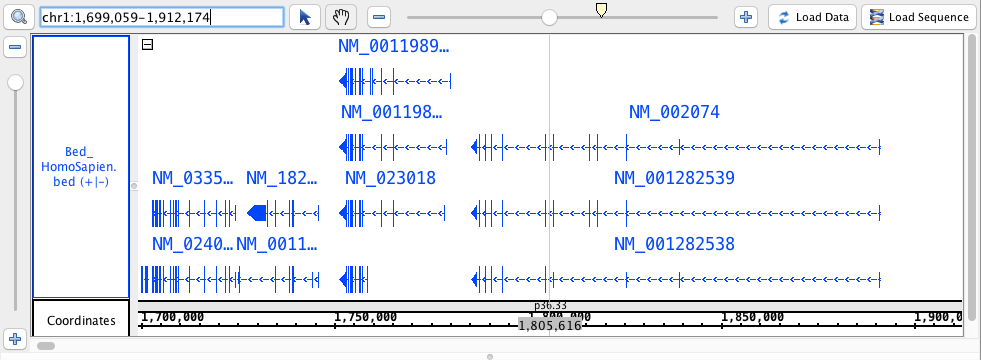
- BedGraph
- BedGraph_HomoSapien.bedgraph
- BedGraph_HomoSapien.bedgraph.gz
chr1:386,893-7,895,983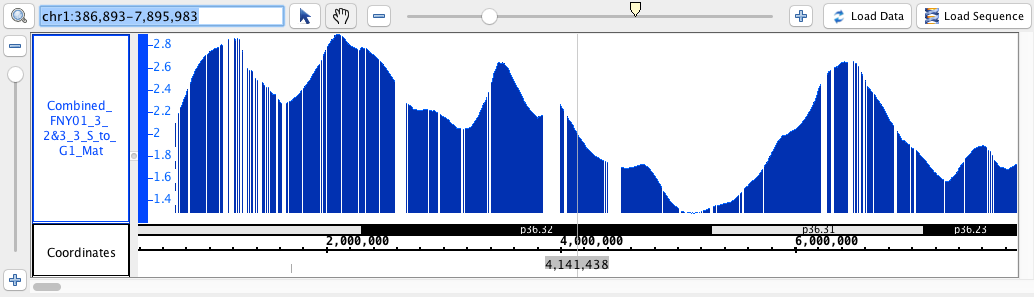
- BigBed
- BigBed_HomoSapien.bigbed
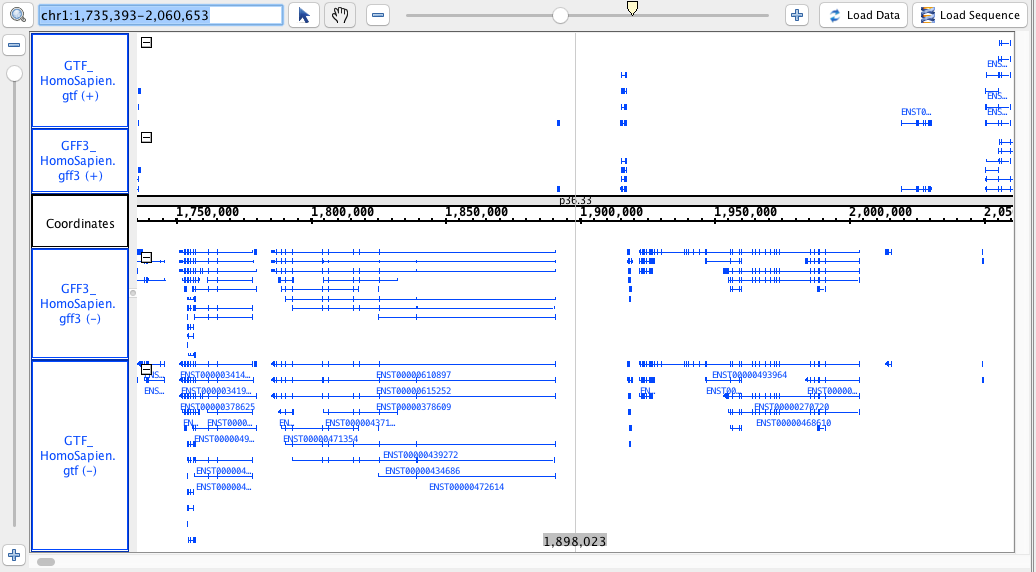
chr1:1,735,393-2,060,653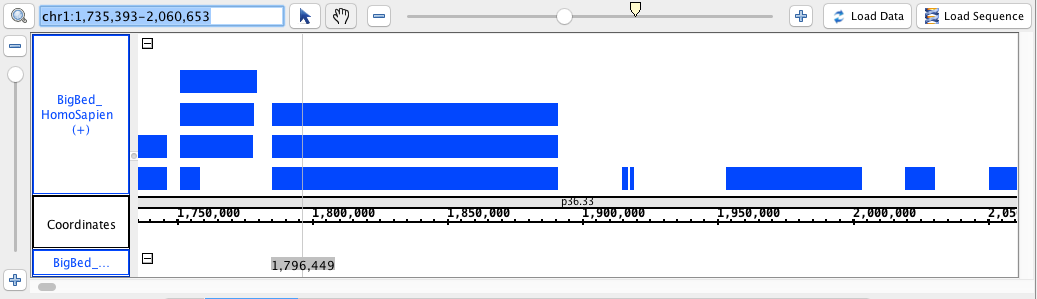
- BigBed_HomoSapien.bigbed
- BigWig
- BigWig_HomoSapien,bigwig
chr1:1,735,393-2,060,653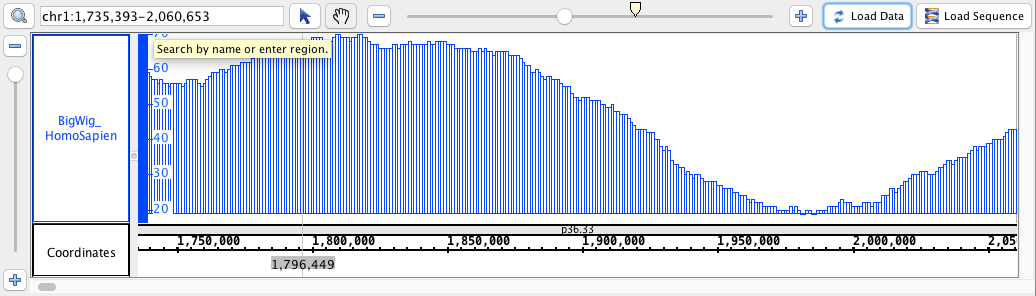
- BigWig_HomoSapien,bigwig
- GFF3
- GFF3_HomoSapien.gff3
- GFF3_HomoSapien.gff3.gz
chr1:1,735,393-2,060,653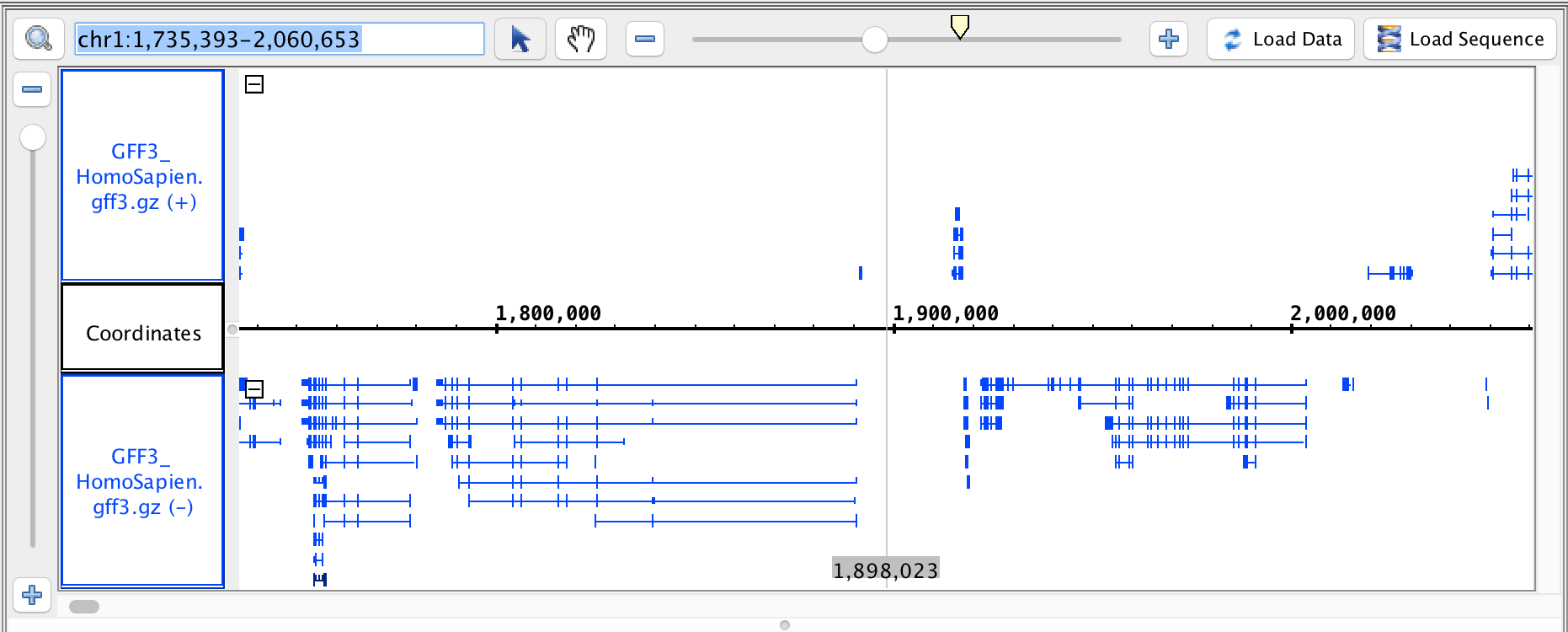
- Bam
- The added Quickload site is available for the Homo sapien genome under the Available Data pane.
- Add a Data Provider to IGB using the following URL: https://quickload-testing.s3.amazonaws.com/smokeTestingQuickload/
- GTF
- GTF_HomoSapien.gtf
- GTF_HomoSapien.gtf.gz
- NarrowAndBroadPeak
- BroadPeak_HomoSapien.broadpeak
- BroadPeak_HomoSapien.broadpeak.gz
chr1:800,118-800,619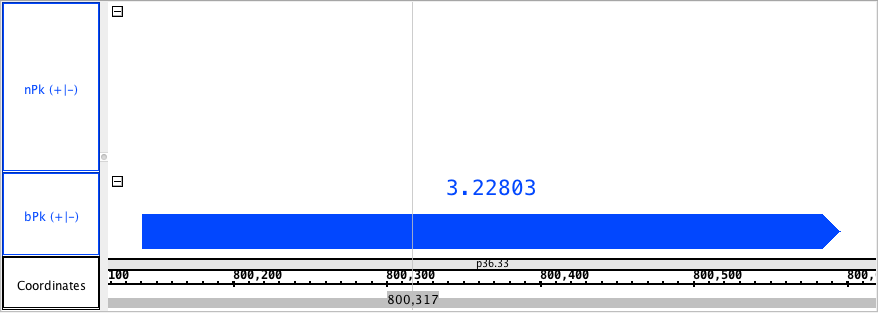
- NarrowPeak_HomoSapien.narrowpeak
- NarrowPeak_HomoSapien.narrowpeak.gz
chr1:9,361,070-9,361,207
chr1:0-12,643,524
- Sam
- Sam_HomoSapien_withHeader.sam
chr1:85,234,475-85,234,620
Load data and sequence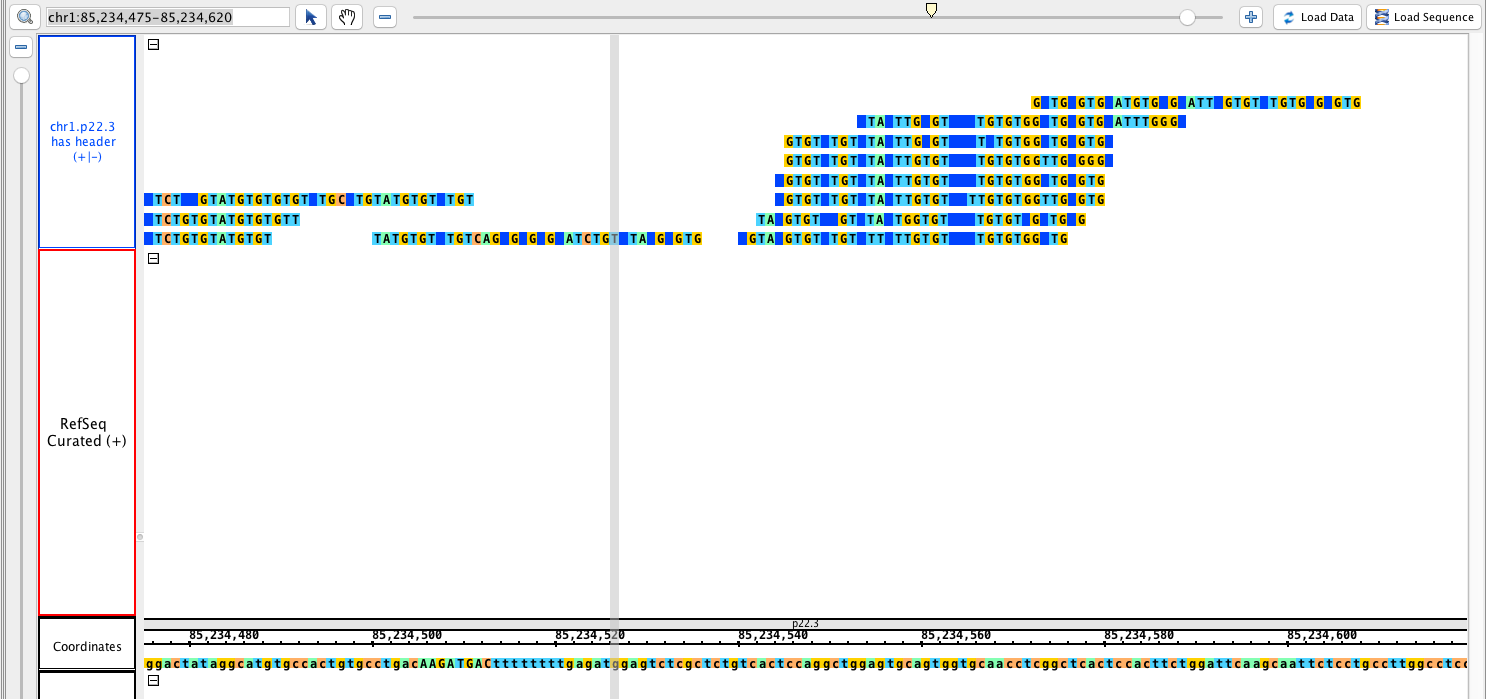
- Sam_HomoSapien_withHeader.sam
- VCF
- VCF_HomoSapien.vcf
- VCF_HomoSapien.vcf.gz
chr1:1,234,555-1,234,601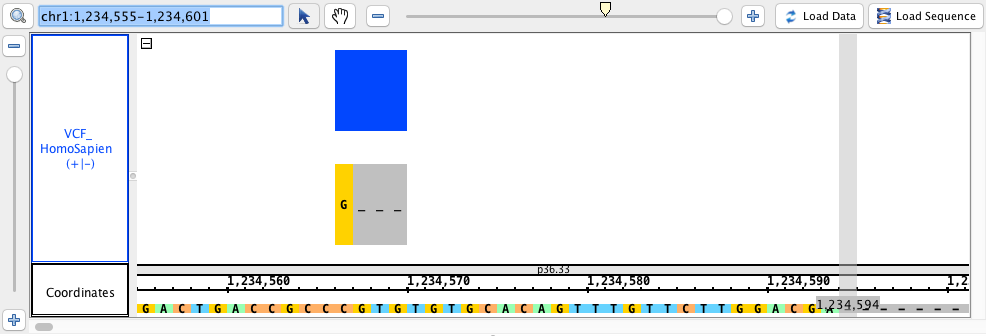
- Wig
- Wig_HomoSapien.wig
- Wig_HomoSapien.wig.gz
chr1:37,750,880-38,042,899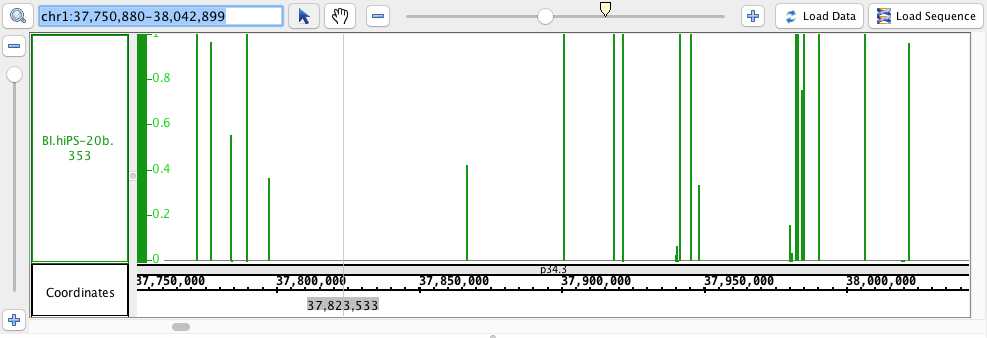
- GTF
Sequence appearance
Go to the Arabidopsis genome. From the RNA-Seq quicklaod (available by default as of 9.0.1), open the file RNA-Seq / Pollen SRP022162 / Reads / Pollen alignments. (url http://lorainelab-quickload.scidas.org/rnaseq/A_thaliana_Jun_2009/SRP022162/Pollen.bam)
Go to location: Chr1:7,313,640-7,319,896
Load Data and Load Sequence. Make sure the +/- option is checked for both the TAIR10_mRNA track (loaded by default) and the Pollen alignments track.
Go to location: Chr1:7,319,301-7,319,375
Optimize the stack height, and compare the main view to this image:
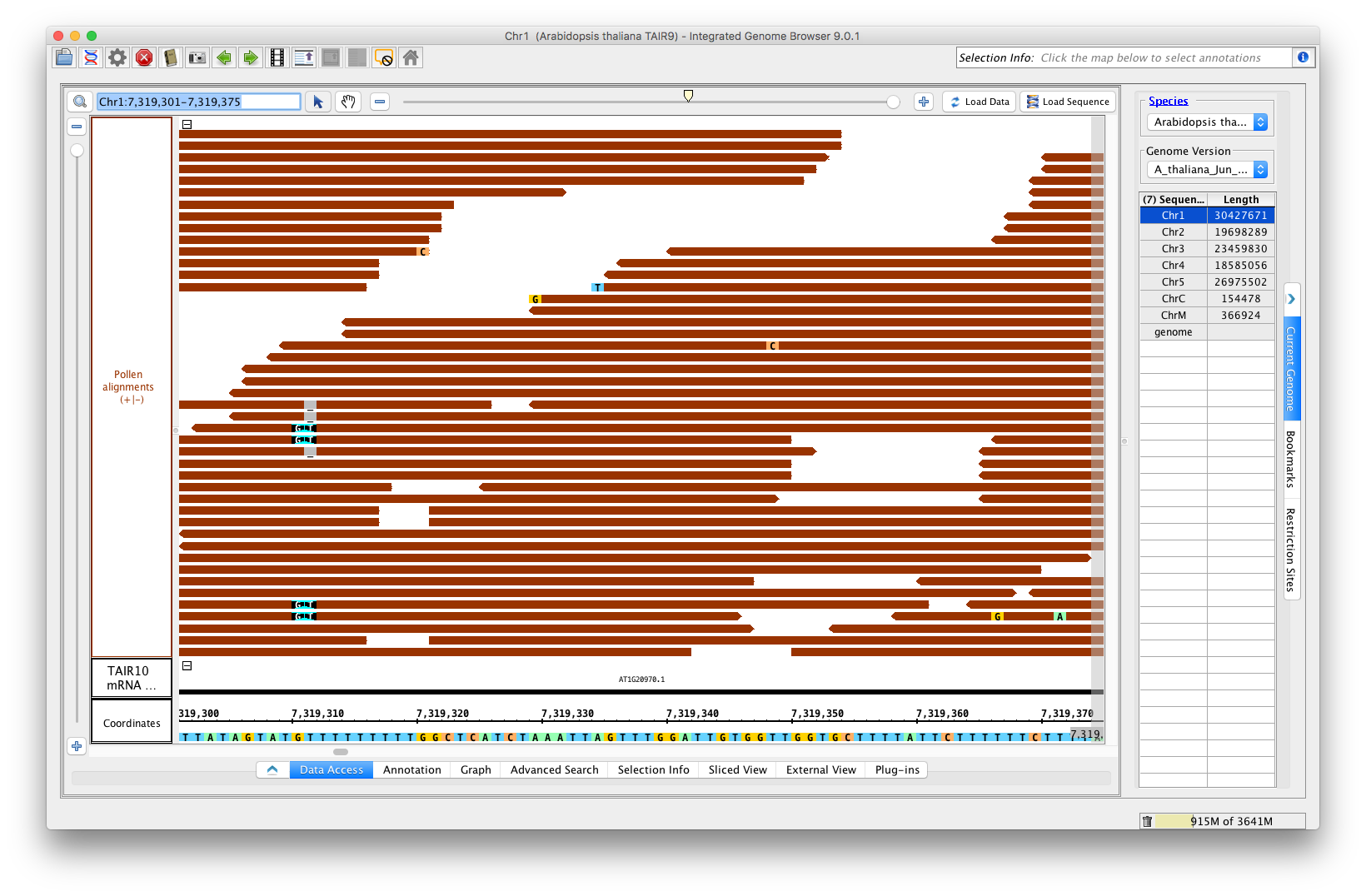
Verify that:
- single base mismatches of A, T, C, and G appear with a color scheme that matches the sequence in the coordinate axis (the bottom).
- deletions appear as shown, a gray space with a dash.
- insertions appear as shown (see position 7,319,310 in image)