| Anchortoc | genandchr | genandchr |
|---|
...
Genomes and
...
All data viewed in IGB, regardless of its source, is organized into distinct genomes and chromosomes.
...
chromosomes
Chromosomes, also called contigs or reference sequences, refer to one or more annotated sequences that form a genome assembly. For completed genome projects, these correspond to the sequence of a physical chromosome. At other times it For less complete genomes, they may represent an assembled contig, a BAC, or any other DNA sequence. All chromosomes in IGB are assumed to be DNA, rather than RNA sequences.A genome version refers assembled contigs that correspond to parts of a physical chromosome.
Genome, genome version, or assembly refer to a group of chromosome sequences that you or another group assembled and made available.
For example, NCBI versions 35 and 36 of the human genome are considered to be two separate genomes. Each one contains multiple chromosome sequences, including the expected chromosomes 1 to 22, X and Y. Other sequences, such as "chr22_random" are also considered distinct chromosomes for the purposes of display in IGB.
Each sequence in IGB is identified by its genome and chromosome names, which must therefore be distinct. No two chromosomes within the same genome version can have the same name.
Naming a Genome
If you are building a genome for display in IGB, we recommend that you give it an IGB-friendly name, consisting of the month and year of release combined with genus and species, following the pattern: G_species_mon_yyyy, where G is the first letter of the genus, mon is the three-letter English abbreviation for the month the genome was released, and year is the year of the release.
For example:
- A_thaliana_Jun_2009
- A_mellifera_Jan_2005
- H_sapiens_Feb_2009
Using this scheme will ensure that IGB displays the latest genome first in the genome menu under the Data Access tab.
Adding a Common Name for a Species
When users operate the pulldown menu to choose a species to view in IGB, a 'tool tip' message indicating the common name of the species appears. If you are adding a new species, contact the IGB developers and ask to have your common name added to the species.txt file under version control at sourceforge.net. This is a tab delimited file that lists all the species that IGB supports, including common names for many of them.
Synonyms
Unfortunately, different groups tend to refer to the same genome or chromosome by different names. For example, NCBI human genome build 35 is also known as hg17 and ensembl1834, as well as H_sapiens_May_2004. When IGB is able to recognize that two names refer to the same genome or chromosome, it will merge the data. Otherwise it will keep the two data sets distinct. Currently, IGB uses a simple table of synonyms to store these associations. You can create your own set of synonyms that will extend this set if needed.
Annotations, Sequences, Graphs and Alignments
IGB can work with four distinct types of data: annotations, alignments (typically from Illumina sequencing experiments), graphs and genomic sequences. Some features of the program are type-specific and will only work with specific types of data.
annotated sequences corresponding to the genome sequence of an organism. IGB designates these using the month and year they were published or made publicly available.
...
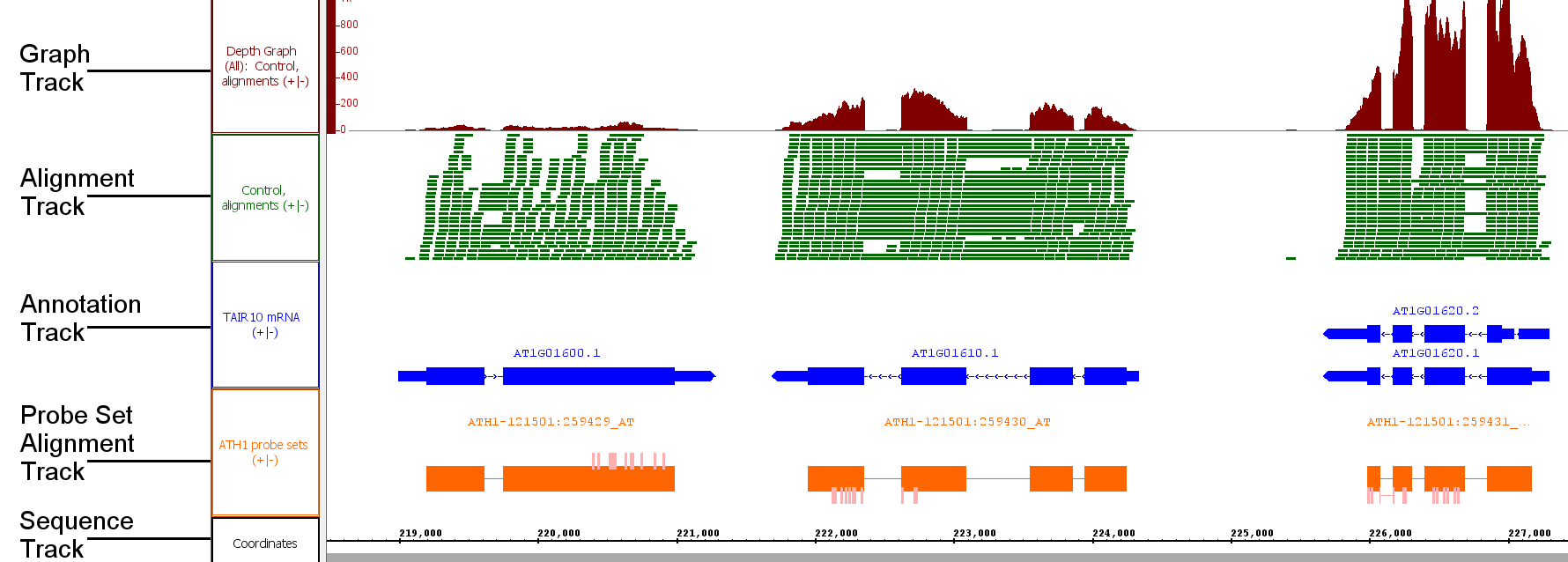
Tracks
Tracks are rows of data read from the same file or data set. When you open a file, the data within the file will appear in one or more tracks. Older versions of IGB referred to tracks as "tiers" and so you may see this term used elsewhere in the User's guide. There are three main types of tracks: Graph tracks, Annotation tracks, and Reference Sequence tracks. Alignment tracks and Probe Set tracks are types of Annotation Track.
...
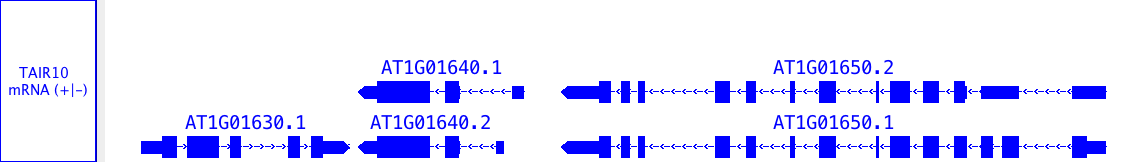
Annotation tracks
Annotations indicate the known or suspected locations of genomic landmark features such as genes, exons, promoter regions, pseudogenes, and so forth. Alignments of EST sequences, GeneChip probe sequences, and other sequences onto chromosome are also sometimes referred to as annotations, particularly when they don't include the sequence of the aligned entity. Annotation data can be loaded from files, QuickLoad and DAS servers.
Sequences are sets of DNA residues comprising a chromosome. Sequences can be loaded from files, QuickLoad, and DAS servers. It is recommended to load sequence data only for small regions of the genome at a time.
Graphs indicate scores or other numeric values as a function of genomic position. Graphs are generally displayed as some form of plot (x,y-plot, bar plot, etc.). The results from tiling arrays are generally represented as graphs. There are two types of graph data: point-based graphs, in which numerical values are associated with individual (single) base positions; and interval graphs, which capture values associated with ranges of genomic positions.
Alignments represent how empirical sequences (such as short Annotations may consist of a single coordinate, a single span with a start and end positions, or a collection of spans. Most annotations reside on either the plus or minus strand of a chromosome, but some do not.
Examples of annotations include:
- single-coordinate feature: splice site
- single-span feature, with strand: an exon
- single-span feature, no strand: sequence recognized by a restriction enzyme
- multi-span feature, with strand: a gene model
...
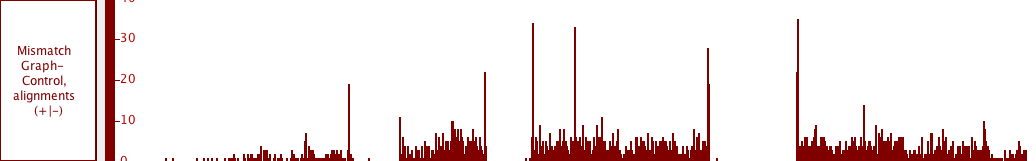
Graphs tracks
Graphs are numeric data associated with regions or single-base positions in a chromosome.
...
Sequence track
Sequences are DNA residues from a chromosomes or contigs that together make an assembly. Sequences can be fully or partially loaded from local files, Quickload sites, or Distributed Annotation Servers. You can view sequence data in the Coordinates track or by opening the Sequence Viewer.
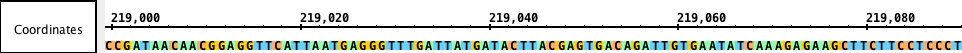
...
Alignments
Alignments represent how sequences obtained from an experiment (such as sequence reads from an RNA-Seq experiment) align onto the a reference genomic sequence. At At low zoom they look like regular annotations, but with marks representing mismatches whenever these data are available. At high zoom they show the sequence of the aligned read, and sometimes indicate scores and the degree of agreement with the reference sequence. These are typically loaded from BAM (binary alignment) files., insertions, or deletions. At higher zoom, the aligned sequence bases become visible.

...
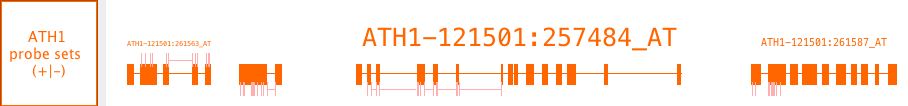
Probe set alignments
Probe set alignments consist of Affymetrix probe set target sequences aligned onto the reference with probe locations indicated as annotations on the target sequence alignments. The probes are kind of annotation on an annotation.
These data are Affymetrix specific.