MetaCerberus is a massively parallel, fast, low memory, scalable annotation tool for inference gene function across genomes to metacommunities. It offers scalable gene elucidation to major public databases, including KEGG (KO), COGs, CAZy, FOAM, and specific databases for viruses, including VOGs and PHROGs, from single genomes to metacommunities. More information about the tool as well as its source code can be found on its GitHub page: https://github.com/raw-lab/MetaCerberus. Below, you'll find the the process for visualizing MetaCerberus output in IGB.
Install MetaCerberus
MetaCerberus is compatible with Python 3, works on both Mac OS X and Linux, and can be installed using bioconda:
Linux/OSX-64
1) Install mamba using conda
conda install mamba
NOTE: Make sure you install mamba in your base conda environment unless you have OSX with ARM architecture (M1/M2 Macs). Follow the OSX-ARM instructions below if you have a Mac with ARM architecture.
2) Install MetaCerberus with mamba
mamba create -n metacerberus -c conda-forge -c bioconda metacerberus conda activate metacerberus metacerberus.py --setup metacerberus.py --download
OSX-ARM (M1/M2)
1) Set up conda environment
conda create -y -n metacerberus conda activate metacerberus conda config --env --set subdir osx-64
2) Install mamba, python, and pydantic inside the environment
conda install -y -c conda-forge mamba python=3.10 "pydantic<2"
3) Install MetaCerberus with mamba
mamba install -y -c conda-forge -c bioconda metacerberus metacerberus.py --setup metacerberus.py --download
NOTE: Mamba is the fastest installer. Anaconda or miniconda can be slow. Also, install mamba from conda not from pip. The pip mamba doesn't work for install.
Open MetaCerberus output in IGB
Run MetaCerberus
Run metacerberus.py with the options required for your project. See MetaCerberus' GitHub page for usage details: https://github.com/raw-lab/MetaCerberus?tab=readme-ov-file#metacerberus-options
Add custom genome
Before you can visualize the MetaCerberus output, the genome used to run MetaCerberus will need to be added as a custom genome in IGB.
However, if your genome is not available, you can still use IGB. Here's how:
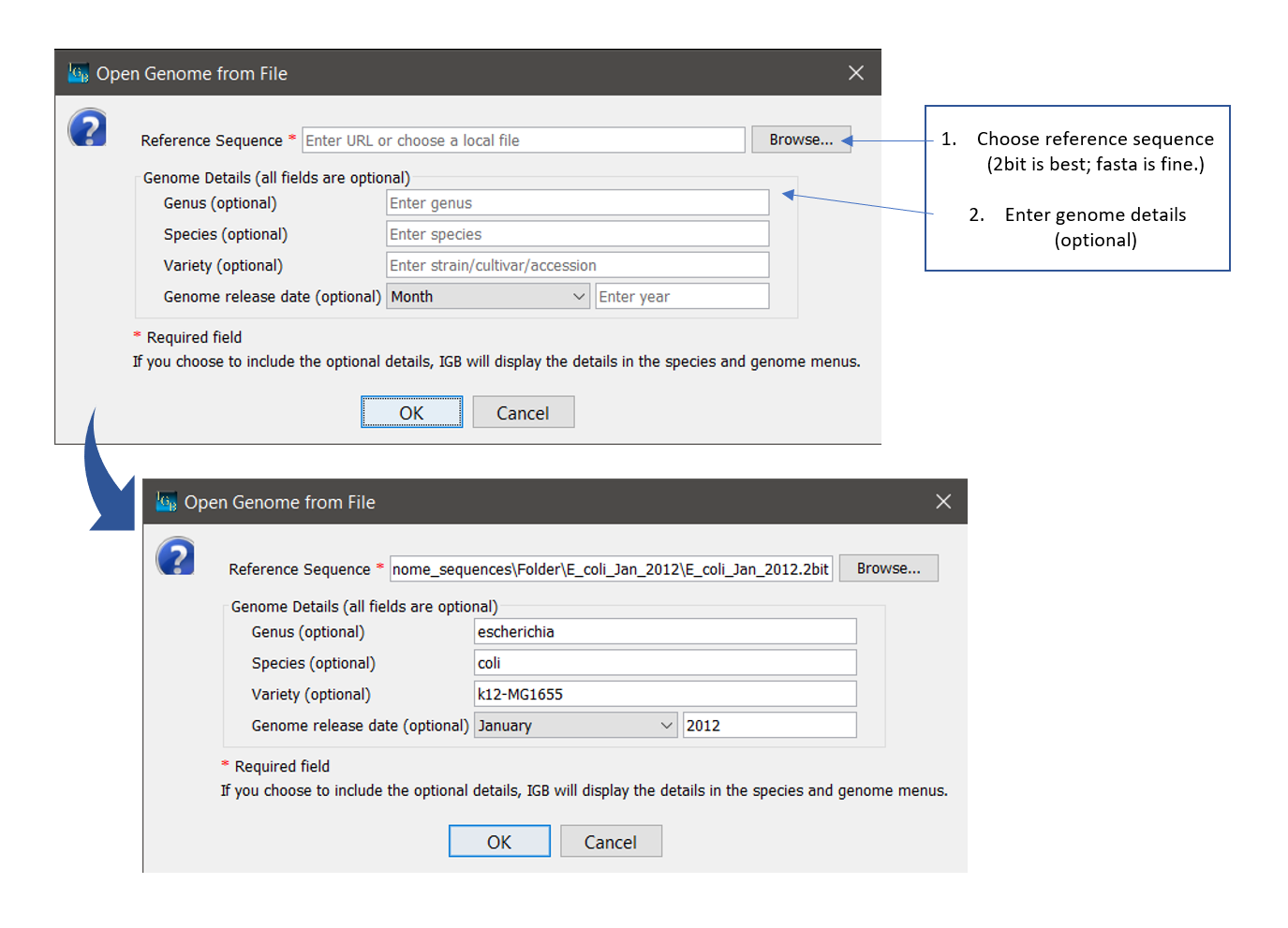
How to open a custom genome
- Select File > Open Genome from File... (or click the DNA icon in the Toolbar.)
- Select a sequence file to use as the reference genome (fasta or 2bit format).
- Enter Optional details:
- Enter Genus name
- Enter the Species name
- Enter the Variety as appropriate (strain/cultivar/accession)
- Choose the Month of the genome release date
- Enter the Year in YYYY format.
- Click OK and wait for the genome to load.
- Open data files as usual. And to view sequence, zoom in and click Load Sequence.
Visualize output
Output files with a .gff extension can be viewed in IGB. See File Formats for a list of all currently supported file formats.
To open local files on your computer:
- Select File > Open File... or File > Open URL...
- Enter file name or URL
Alternatively, drag and drop local files from your file chooser into IGB.

Some MetaCerberus output in the .gff file format will have a ##FASTA section at the bottom. Although this is a valid component of a .gff file, IGB does not currently have the logic to parse it and has thus been throwing an error when it encounters files with this section. We are working on adding that logic, but in the meantime, removing all text in your .gff file from that ##FASTA section down will fix the issue.