Bioviz Connect utilizes CyVerse’s cloud computing to perform powerful analyses on genomic data.
To Analyze:
To analyze a file right click and choose ‘Analyse’. A panel will open on the right with the available analyses, if there are any. Click the appropriate button for the analysis you want to run.
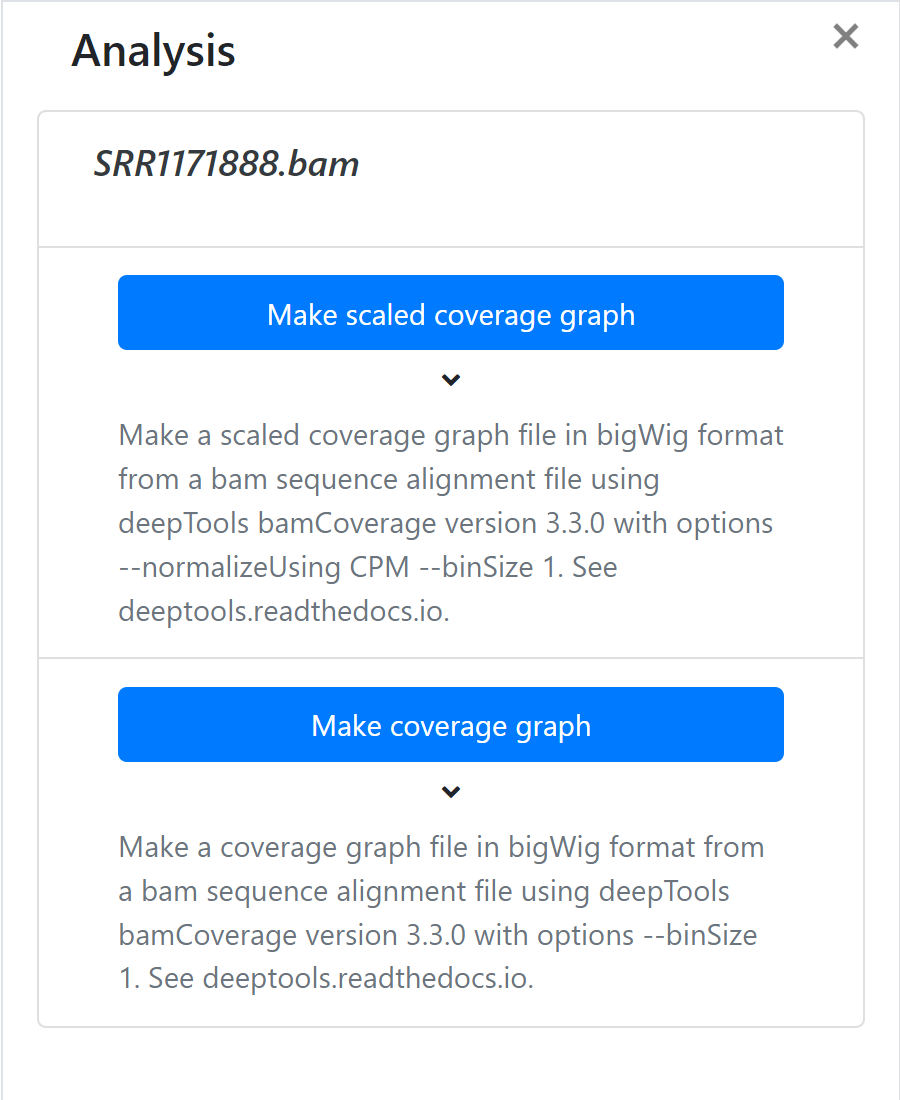

Once you have chosen a type of analysis, a new form will appear in the same window. Enter your analysis name. An output file name will be generated for you. This can be edited to any name you like.
When you have completed entering your information click 'Run Analysis'.
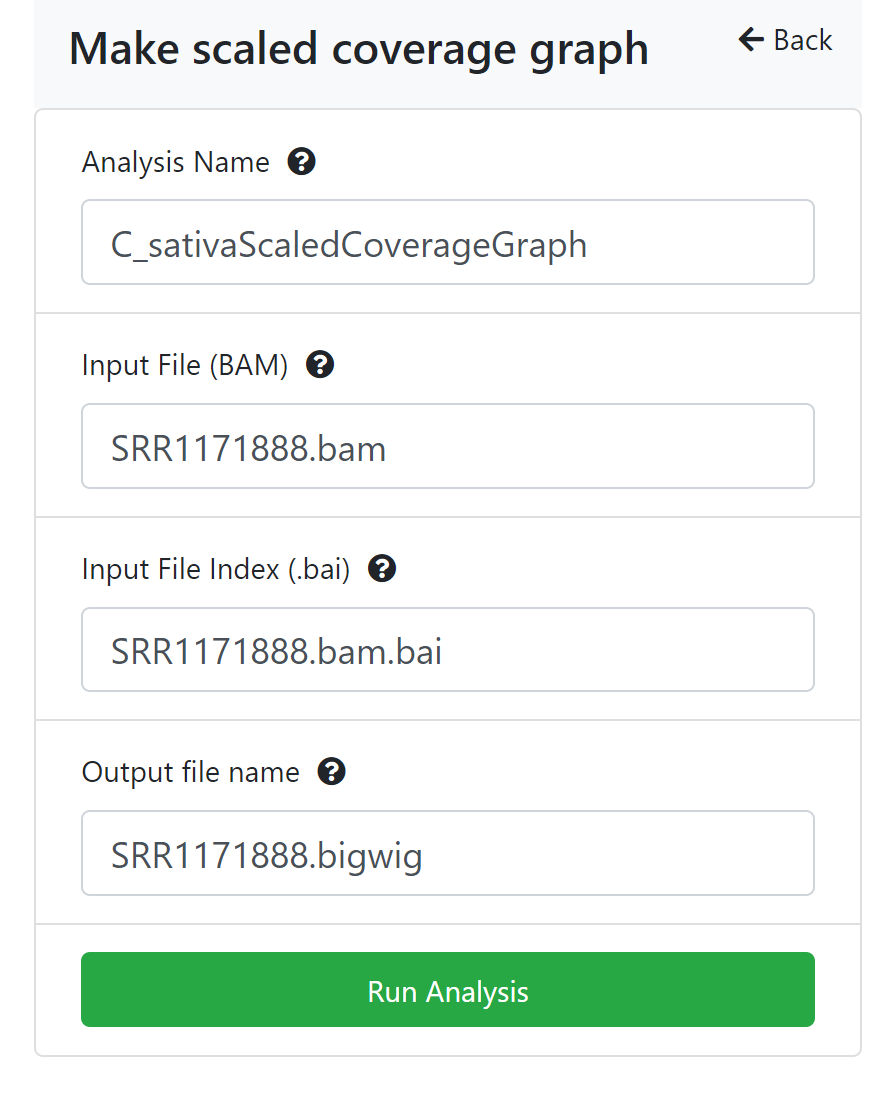
Analysis Status
You can check the analyses’ status by using the ‘Analyses History’ tab, where analyses are listed as Queued (waiting to run), Running, Failed, or Completed. The length of the time to complete a job is dependent on the size of the queue, the analysis being carried out, and the size of the file. It will likely take several minutes for the analysis to complete. CyVerse will send an email to you when the analysis is finished.
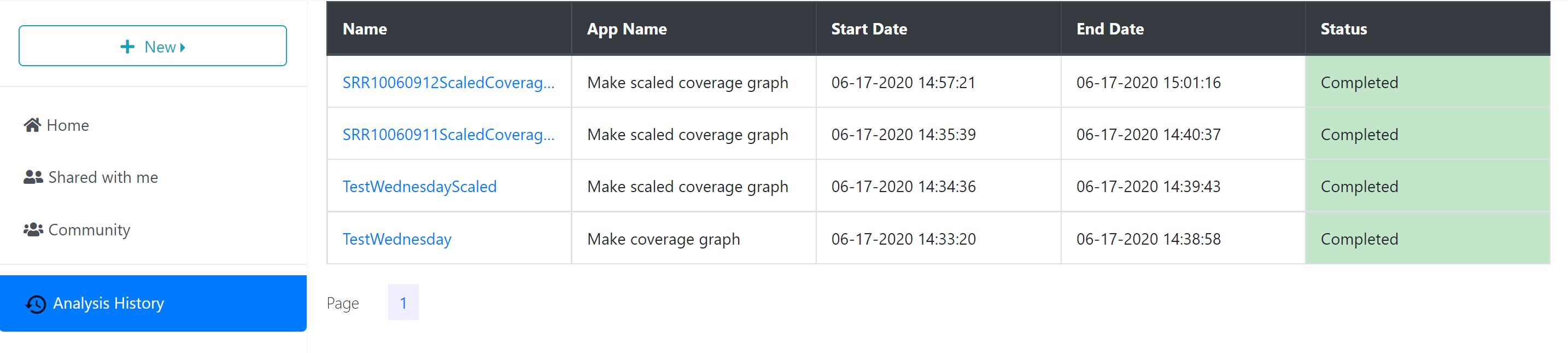
Analysis Results
When the analysis is finished, any files or folders created through the process will appear in the ‘Analyses’ folder in your ‘Home’ directory, or in the same location as the input files if those are stored in a location where you have permission to modify or add to the folder.
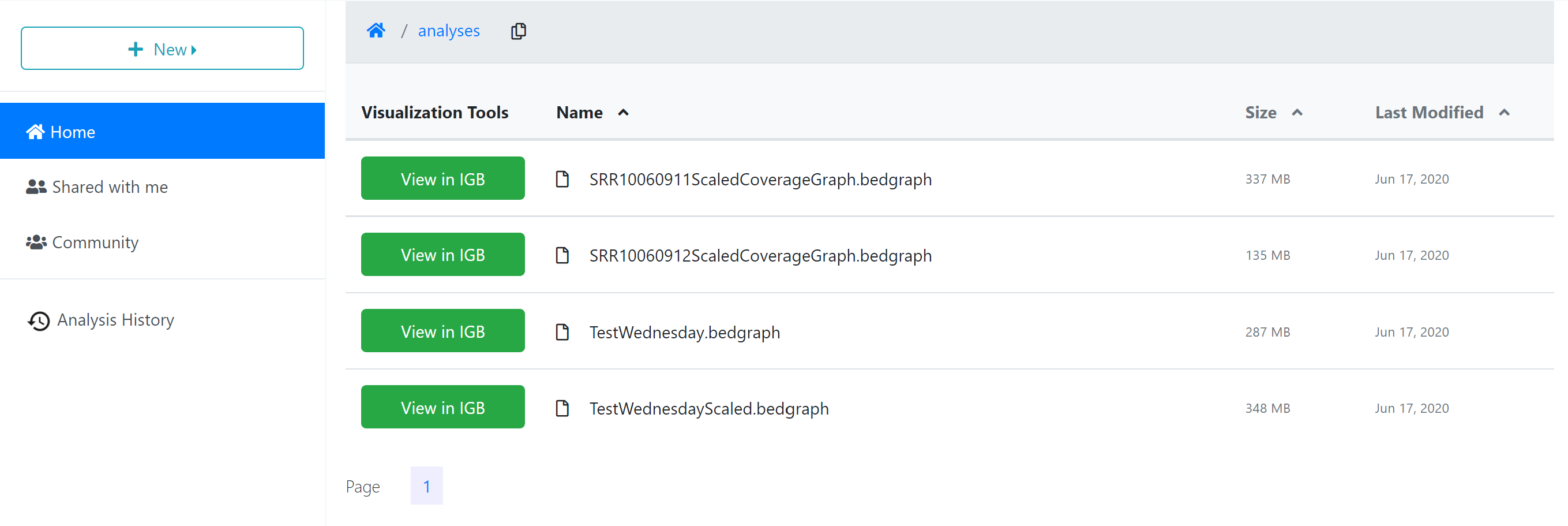
To quickly navigate to the analysis results, you can click the analysis name in the ‘Analyses History’ tab, opening the folder where the output data files are stored.
Note that running analyses uses Compute Units which may be limited by your account’s subscription status. See cyverse.org/ for more information.
Supported File Types
Currently BioViz Connect supports analyses of:
- bam files
- genome wide coverage graph
- Make a coverage graph file in bigWig format from a bam sequence alignment file using deepTools bamCoverage version 3.3.0 with options --binSize 1. See deeptools.readthedocs.io.
- genome wide scaled coverage graph
- Make a scaled coverage graph file in bigWig format from a bam sequence alignment file using deepTools bamCoverage version 3.3.0 with options --normalizeUsing CPM --binSize 1. See deeptools.readthedocs.io.
- fasta files
- Find enriched sequence motifs
- Find overrepresented sequences in a fasta file using DREME from meme-suite.org. DREME discovers short, ungapped motifs that are relatively enriched in your sequences compared with shuffled sequences.